Advanced Ofloxacin Manufacturing: Dual-Protection Strategy for High-Yield API Production
Advanced Ofloxacin Manufacturing: Dual-Protection Strategy for High-Yield API Production
The pharmaceutical industry continuously seeks robust synthetic routes that maximize yield while minimizing impurity profiles, particularly for high-volume fluoroquinolone antibiotics like Ofloxacin. Patent CN103360410A introduces a transformative preparation method that addresses longstanding inefficiencies in the traditional seven-step synthesis originating from tetrafluorobenzoic acid. This innovation centers on a novel dual-protection strategy utilizing trimethylchlorosilane, which simultaneously safeguards both hydroxyl and amino functional groups during the critical acylation phase. By streamlining what was previously a multi-step protection and deprotection sequence into a more efficient single operational flow, this technology significantly enhances the utilization degree of the expensive acylating agent, (2,3,4,5)-tetrafluorobenzoyl chloride. For R&D directors and process chemists, this represents a pivotal shift towards greener, more atom-economical manufacturing, offering a reliable pathway to high-purity API intermediates with reduced waste generation.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial production of Ofloxacin and its levo-isomer has relied on routes that are chemically cumbersome and prone to yield erosion. The conventional seven-step synthesis starting from tetrafluorobenzoic acid often suffers from incomplete reactions where unreacted intermediates readily engage in side reactions with aminopropanol, leading to complex impurity profiles that are difficult to purge. These side reactions not only compromise the purity of the final active pharmaceutical ingredient but also necessitate rigorous and costly purification steps, such as extensive chromatography or repeated recrystallizations. Furthermore, the traditional separation of protection steps for hydroxyl and amino groups adds unnecessary operational complexity, extending cycle times and increasing solvent consumption. For supply chain managers, these inefficiencies translate into volatile production schedules and higher raw material costs, as the poor utilization of key reagents like tetrafluorobenzoyl chloride drives up the cost of goods sold.
The Novel Approach
The methodology disclosed in CN103360410A fundamentally reengineers the early stages of the synthesis to overcome these bottlenecks. By employing trimethylchlorosilane in the presence of a Lewis base catalyst, the process achieves simultaneous protection of both the hydroxyl and amido functionalities in a single pot operation. This dual-protection mechanism effectively masks reactive sites that would otherwise lead to polymerization or unwanted acylation, ensuring that the subsequent reaction with (2,3,4,5)-tetrafluorobenzoyl chloride proceeds with high chemoselectivity. The result is a dramatic reduction in side product formation and a notable increase in the yield of the critical difluorocarboxylic acid intermediate, reported to improve by approximately 10 percent compared to commercial baselines. This approach not only simplifies the workflow by eliminating intermediate isolation steps but also maximizes the value derived from every kilogram of starting material, offering a compelling value proposition for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Trimethylchlorosilane Dual-Protection and Cyclization
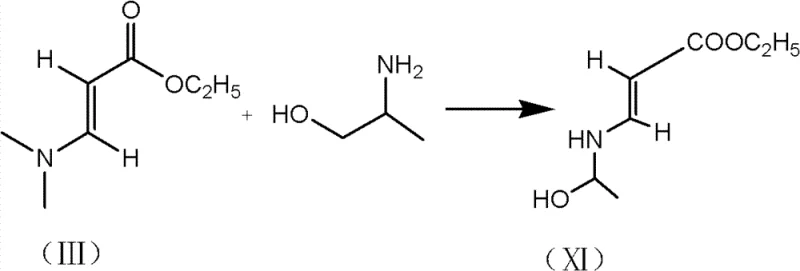
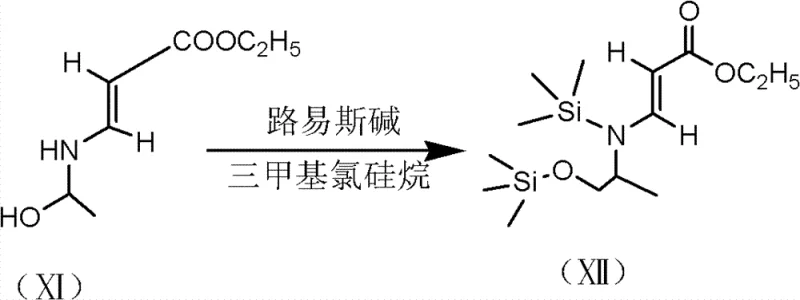
The core innovation of this process lies in the mechanistic behavior of trimethylchlorosilane (TMSCl) under Lewis base catalysis. In standard organic synthesis, protecting groups are often applied sequentially, requiring distinct reaction conditions and workup procedures for each functional group. However, in this specific protocol, the Lewis base—selected from options such as triethylamine, liquefied ammonia, or DMF—activates the silane reagent to rapidly silylate both the alcohol and the amine moieties of the aminopropanol derivative. This creates a robust bis-silylated intermediate (Compound XII) that is sterically hindered and electronically deactivated towards nucleophilic attack at the nitrogen and oxygen sites. When (2,3,4,5)-tetrafluorobenzoyl chloride is introduced, it reacts exclusively at the intended carbon center without interference from the protected groups. This precise control over reactivity is crucial for maintaining high purity, as it prevents the formation of N-acylated or O-acylated byproducts that typically plague fluoroquinolone synthesis. The subsequent acid washing step efficiently cleaves these silyl groups, regenerating the free hydroxyl and amine functions ready for the intramolecular cyclization.
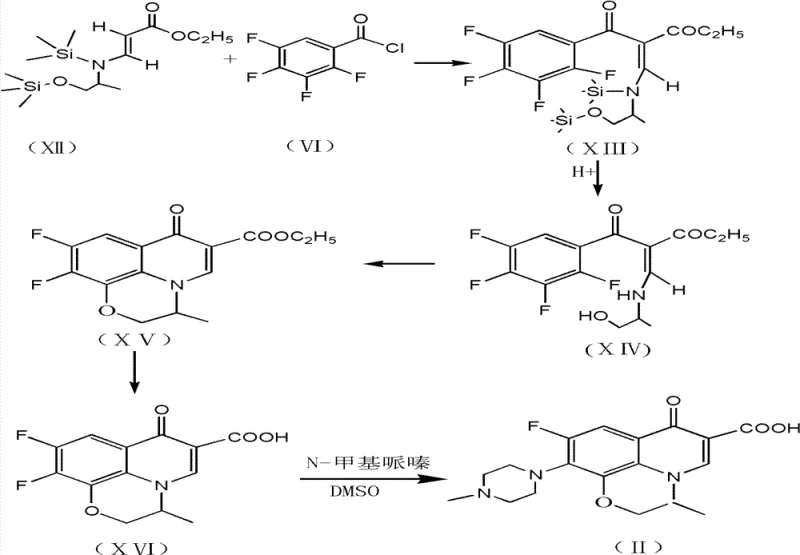
Following the acylation and deprotection, the synthesis proceeds through a critical cyclization step mediated by anhydrous potassium fluoride in dimethyl formamide (DMF). The fluoride ion acts as a potent nucleophile, facilitating the displacement of the ortho-fluorine atom on the benzoyl ring by the secondary amine, closing the quinolone core. This step is performed under reflux conditions, typically between 90°C and 100°C, ensuring complete conversion to the cyclic ester. The use of anhydrous conditions is paramount here to prevent hydrolysis of the ester prior to cyclization. After the ring is formed, acid-catalyzed hydrolysis converts the ester to the carboxylic acid, yielding the difluorocarboxylic acid precursor. Finally, the displacement of the remaining fluorine atom by N-methyl piperazine in DMSO completes the molecule. This sequence demonstrates a high degree of process intensification, where reaction conditions are optimized to drive equilibrium towards the desired product while minimizing thermal degradation.
How to Synthesize Ofloxacin Efficiently
The implementation of this dual-protection strategy requires careful attention to stoichiometry and temperature control to replicate the high yields reported in the patent data. The process begins with the condensation of (N,N)-dimethylamino ethyl acrylate and aminopropanol in toluene, followed by the in-situ addition of the silylating agents. Operators must maintain the reaction temperature between 40°C and 50°C during the protection phase to ensure complete silylation without decomposing the sensitive enamine structure. Once the protected intermediate is formed, the acylation with tetrafluorobenzoyl chloride is conducted at elevated temperatures (50-100°C) to drive the reaction to completion. The subsequent cyclization in DMF with potassium fluoride is the most critical step for yield determination; it requires strict exclusion of moisture and precise reflux management. Detailed standardized operating procedures for scaling this chemistry from laboratory benchtop to commercial production are essential for maintaining batch-to-batch consistency.
- React (N,N)-dimethylamino ethyl acrylate with aminopropanols in methylbenzene, followed by protection with trimethylchlorosilane and Lewis base.
- Perform acylation with (2,3,4,5)-tetrafluorobenzoyl chloride, followed by acid washing to remove protecting groups.
- Execute cyclization using anhydrous potassium fluoride in DMF, hydrolyze to difluorocarboxylic acid, and condense with N-methyl piperazine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers tangible strategic advantages beyond mere technical elegance. The primary economic driver is the substantial improvement in raw material efficiency, specifically regarding the utilization of (2,3,4,5)-tetrafluorobenzoyl chloride, which is a high-cost specialty chemical. By mitigating side reactions through effective dual protection, the process ensures that a greater proportion of this expensive reagent is incorporated into the final product rather than being lost to waste streams or byproduct formation. This directly translates to a lower cost per kilogram of the API intermediate, providing a competitive edge in pricing negotiations. Furthermore, the reduction in impurity generation simplifies the downstream purification train, potentially reducing the volume of solvents and adsorbents required for polishing, which aligns with broader sustainability goals and waste disposal cost reductions.
- Cost Reduction in Manufacturing: The elimination of separate protection and deprotection steps significantly shortens the overall production cycle time, reducing labor and utility costs associated with extended reactor occupancy. Additionally, the increased yield of the difluorocarboxylic acid intermediate means that less starting material is required to produce the same amount of final API, effectively lowering the variable cost of production. The process avoids the use of transition metal catalysts that often require expensive removal steps, further streamlining the cost structure and enhancing the overall economic viability of large-scale manufacturing operations.
- Enhanced Supply Chain Reliability: By simplifying the synthetic route and reducing the number of unit operations, the process becomes inherently more robust and less susceptible to variability, ensuring consistent supply continuity for downstream formulation partners. The reagents used, such as trimethylchlorosilane and common Lewis bases, are widely available commodity chemicals, reducing the risk of supply bottlenecks associated with exotic or proprietary catalysts. This reliability allows for more accurate demand forecasting and inventory planning, minimizing the need for safety stock and freeing up working capital for other strategic investments within the supply chain network.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing standard solvents like toluene and DMF that are easily recovered and recycled in modern chemical plants, thereby minimizing environmental impact and regulatory compliance burdens. The reduction in waste generation, driven by higher selectivity and yield, lowers the burden on effluent treatment facilities and reduces the carbon footprint of the manufacturing site. This alignment with green chemistry principles not only satisfies increasingly stringent environmental regulations but also enhances the corporate social responsibility profile of the manufacturing partner, making it a preferred vendor for global pharmaceutical companies with strict sustainability mandates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this dual-protection synthesis route for Ofloxacin. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing a factual basis for evaluating the technology's fit within your existing manufacturing portfolio. Understanding these nuances is critical for R&D teams assessing process transfer feasibility and for procurement teams evaluating the long-term cost implications of adopting this new methodology.
Q: How does the trimethylchlorosilane protection improve the Ofloxacin synthesis yield?
A: The use of trimethylchlorosilane simultaneously protects both hydroxyl and amino groups, preventing side reactions during acylation and increasing the utilization efficiency of (2,3,4,5)-tetrafluorobenzoyl chloride, thereby boosting the intermediate yield by approximately 10%.
Q: What are the critical reaction conditions for the cyclization step?
A: The cyclization involves adding anhydrous potassium fluoride to a DMF solution at reflux temperatures. The process requires precise temperature control, typically maintaining the reaction mixture under reflux for several hours to ensure complete ring closure before hydrolysis.
Q: Can this process be adapted for Levofloxacin production?
A: Yes, the process is stereospecific. By substituting L-aminopropanol with D-aminopropanol in the initial condensation step, the same synthetic route effectively produces Levofloxacin, demonstrating the versatility of this dual-protection methodology.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ofloxacin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient, high-yield synthetic routes in the competitive landscape of generic antibiotic production. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovations like the trimethylchlorosilane dual-protection method can be seamlessly transferred from pilot scale to full industrial output. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of Ofloxacin or Levofloxacin intermediate meets the highest international pharmacopoeial standards. Our commitment to quality assurance ensures that the impurity profiles remain well within acceptable limits, safeguarding the safety and efficacy of the final drug product.
We invite global pharmaceutical partners to collaborate with us to leverage this advanced manufacturing technology for their supply chains. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis that quantifies the potential economic benefits of switching to this optimized route for your specific volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that enhance your operational efficiency and market competitiveness in the fluoroquinolone sector.