Revolutionizing Hydroxychloroquine Sulfate Production: A High-Yield Industrial Breakthrough
Revolutionizing Hydroxychloroquine Sulfate Production: A High-Yield Industrial Breakthrough
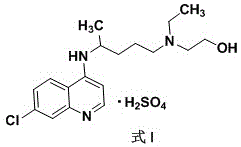
The pharmaceutical industry constantly seeks robust, scalable, and high-purity synthetic routes for critical active pharmaceutical ingredients (APIs). A significant advancement in this domain is detailed in patent CN111606851A, which discloses an industrial preparation method for hydroxychloroquine sulfate characterized by exceptional purity and yield. This technology addresses long-standing challenges in the condensation of quinoline derivatives, offering a pathway that operates under mild conditions without the need for complex inert atmospheres. By shifting the reaction paradigm from harsh, high-temperature solvent-free environments to controlled alcoholic solvent systems, this innovation ensures that the final product meets stringent pharmacopeial standards with a purity exceeding 99.80% and a maximum single impurity content of less than 0.10%. For global supply chains, this represents a pivotal shift towards more reliable and cost-effective manufacturing of this vital antimalarial and immunomodulatory agent.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of hydroxychloroquine has been plagued by severe process limitations that hindered large-scale efficiency and product quality. Prior art, including various patents and literature, typically relies on condensation reactions conducted at excessively high temperatures, often exceeding 120°C and sometimes reaching up to 130°C. These aggressive thermal conditions create a precarious balance; if the temperature is too low, the conversion of the starting material, 4,7-dichloroquinoline, remains incomplete, leaving significant amounts of unreacted precursor. Conversely, extending the reaction time or increasing the temperature beyond 125°C triggers detrimental side reactions, specifically the formation of deethylation products and nitrogen oxide impurities. Furthermore, some existing methodologies employ solvent-free conditions to drive the reaction, which introduces substantial safety risks due to poor heat transfer coefficients. In large reactors, the disparity between edge and central temperatures can lead to localized overheating, causing batch-to-batch inconsistency and making quality control nearly impossible in an industrial setting.
The Novel Approach
The methodology presented in patent CN111606851A fundamentally reengineers this synthesis by introducing an alcohol-based organic solvent system, preferably ethylene glycol, which acts as both a reaction medium and a thermal buffer. This approach allows the condensation to proceed efficiently at a much milder temperature range of 80-100°C, effectively eliminating the thermal stress that generates impurities in conventional processes. Crucially, this method does not require nitrogen protection, simplifying the operational workflow and reducing the capital expenditure associated with inert gas systems. The use of a solvent ensures uniform heat distribution throughout the reaction mass, preventing the formation of hot spots that degrade product integrity. As a result, the process achieves a hydroxychloroquine yield of not less than 85% with a purity of at least 99.70%, and upon salification, the sulfate salt reaches a purity of 99.80% or higher, demonstrating a clear superiority over the erratic outcomes of high-temperature, solvent-free alternatives.
Mechanistic Insights into Alcohol-Mediated Condensation
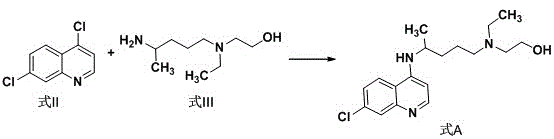
The core of this technological breakthrough lies in the nucleophilic substitution mechanism facilitated by the specific choice of solvent and temperature control. The reaction involves the condensation of 4,7-dichloroquinoline (Formula II) with 5-(N-ethyl-N-2-hydroxyethyl amino)-2-pentylamine (Formula III). In traditional high-energy environments, the kinetic energy is sufficient to break not only the desired bonds but also stable ethyl groups, leading to deethylation. By moderating the thermal energy to the 80-100°C window within a polar alcoholic solvent, the activation energy barrier for the primary substitution is overcome while the barriers for secondary decomposition reactions remain insurmountable. The solvent molecules likely stabilize the transition state through hydrogen bonding interactions, enhancing the nucleophilicity of the amine side chain without promoting its degradation. This precise tuning of reaction parameters ensures that the chlorine atom at the 4-position of the quinoline ring is selectively displaced by the amine, preserving the structural integrity of the ethyl and hydroxyethyl groups essential for the drug's biological activity.
Impurity control is another critical aspect governed by this mechanistic understanding. The patent data indicates that the maximum single impurity is maintained below 0.10%, a testament to the suppression of side pathways. In the absence of excessive heat, the formation of nitrogen oxides is minimized, and the stability of the intermediate species is preserved. The subsequent purification steps, involving alkalization and extraction with halogenated hydrocarbons followed by recrystallization in ester solvents like ethyl acetate, further refine the product profile. This multi-stage purification strategy leverages the differential solubility of the target molecule versus potential byproducts, ensuring that the final API intermediate or salt meets the rigorous demands of modern pharmaceutical regulation. The ability to consistently achieve such low impurity levels without complex chromatographic separations highlights the elegance and practicality of this synthetic design.
How to Synthesize Hydroxychloroquine Efficiently
The implementation of this synthesis route is designed for seamless integration into existing chemical manufacturing infrastructure. The process begins with the precise charging of reactants into a standard reaction vessel, followed by the addition of the alcoholic solvent. The simplicity of the procedure, which avoids the need for specialized high-pressure or inert atmosphere equipment, makes it highly accessible for contract development and manufacturing organizations (CDMOs). The reaction progress is monitored to ensure complete conversion before proceeding to the workup phase, where standard liquid-liquid extraction techniques are employed to isolate the crude product. Finally, a recrystallization step using environmentally friendlier ester solvents polishes the material to the required specification. For detailed operational parameters, stoichiometry, and specific temperature profiles, please refer to the standardized synthesis guide below.
- Combine 4,7-dichloroquinoline and the amine side chain in an alcoholic organic solvent such as ethylene glycol.
- Heat the reaction mixture to a mild temperature range of 80-100°C and maintain stirring until conversion is complete.
- Perform alkaline workup, extract with halogenated hydrocarbons, and purify the resulting oil using ester solvents.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented process translates into tangible strategic benefits that extend far beyond simple chemical yield. The shift to milder reaction conditions inherently reduces the energy consumption required for heating and cooling cycles, leading to a significant reduction in utility costs per kilogram of produced API. Furthermore, the elimination of nitrogen protection requirements simplifies the reactor setup, allowing for faster turnaround times between batches and increased overall equipment effectiveness (OEE). The robustness of the process against thermal degradation means fewer failed batches and less waste generation, directly contributing to cost reduction in pharmaceutical intermediates manufacturing. By minimizing the formation of difficult-to-remove impurities, the downstream purification burden is drastically simplified, reducing the consumption of expensive solvents and filtration media.
- Cost Reduction in Manufacturing: The economic impact of this technology is driven by the elimination of high-energy inputs and the reduction of waste disposal costs associated with failed high-temperature runs. By avoiding the need for solvent-free conditions which pose safety risks and require specialized engineering controls, the capital expenditure for plant modification is minimized. The high yield and purity reduce the need for reprocessing, ensuring that raw materials are converted into saleable product with maximum efficiency. This streamlined approach allows manufacturers to offer competitive pricing without compromising on the quality standards required for regulatory approval.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by complex processes that are sensitive to minor operational deviations. This new method's tolerance for standard atmospheric conditions and its reliance on common, commercially available solvents like ethylene glycol and ethyl acetate mitigate the risk of raw material shortages. The reproducibility of the reaction ensures that every batch meets the same high specifications, reducing the likelihood of supply disruptions caused by out-of-specification results. This reliability is crucial for maintaining the steady flow of essential medicines to the global market, particularly for drugs with high demand volatility.
- Scalability and Environmental Compliance: Scaling chemical processes from the laboratory to the tonnage scale often reveals hidden thermal hazards, but this method's use of a solvent medium ensures safe heat transfer even in large reactors. The reduced formation of nitrogen oxide byproducts aligns with stricter environmental regulations regarding volatile organic compounds and hazardous emissions. Additionally, the use of ester solvents for purification offers a greener alternative to more toxic options, supporting corporate sustainability goals. The process is inherently designed for commercial scale-up of complex pharmaceutical intermediates, ensuring that production can be ramped up quickly to meet surges in demand.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of hydroxychloroquine sulfate using this advanced methodology. These insights are derived directly from the patent specifications and are intended to clarify the operational advantages and quality benchmarks associated with this synthesis route. Understanding these details is essential for partners looking to integrate this technology into their supply chains.
Q: How does the new process control impurities compared to traditional high-temperature methods?
A: Traditional methods operating above 125°C often generate deethylation products and nitrogen oxides. The patented process maintains temperatures below 100°C, significantly suppressing these thermal degradation pathways while ensuring complete conversion.
Q: What are the advantages of using alcoholic solvents over solvent-free conditions?
A: Solvent-free reactions pose significant heat transfer risks, leading to uneven heating and localized hot spots that degrade product quality. Using alcoholic solvents like ethylene glycol ensures uniform heat distribution and safer, more controllable reaction kinetics.
Q: Is nitrogen protection required for this synthesis?
A: No, unlike many sensitive pharmaceutical syntheses, this specific condensation reaction is robust enough to proceed without inert gas protection, simplifying the operational setup and reducing equipment costs.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Hydroxychloroquine Sulfate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting cutting-edge synthetic technologies to ensure the availability of high-quality pharmaceutical ingredients. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from pilot scale to full manufacturing is seamless and efficient. We are committed to maintaining stringent purity specifications and utilizing our rigorous QC labs to verify that every batch of hydroxychloroquine sulfate meets or exceeds the 99.80% purity benchmark established by the latest patents. Our facility is equipped to handle the specific solvent systems and thermal profiles required by this advanced process, guaranteeing a consistent supply of this vital API.
We invite global pharmaceutical partners to collaborate with us to leverage these technological advancements for their product pipelines. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to obtain specific COA data and route feasibility assessments, ensuring that your supply chain is built on the foundation of the most efficient and reliable chemistry available in the market.