Advanced Two-Step Synthesis of Everolimus for Scalable Pharmaceutical Manufacturing
Advanced Two-Step Synthesis of Everolimus for Scalable Pharmaceutical Manufacturing
The pharmaceutical landscape for immunosuppressants continues to evolve, driven by the need for more efficient and economically viable synthetic routes for complex macrolides. A pivotal development in this domain is detailed in Chinese Patent CN102127092B, which outlines a robust preparation method for Everolimus, a next-generation rapamycin analogue widely used in transplant medicine and oncology. This patent addresses critical bottlenecks in existing manufacturing protocols, specifically targeting the low yields and high costs associated with traditional silylation and deprotection sequences. By optimizing solvent systems and reaction parameters, the disclosed methodology offers a pathway to significantly enhance the purity and throughput of this high-value active pharmaceutical ingredient (API). For R&D directors and procurement strategists, understanding the nuances of this improved synthesis is essential for securing a reliable Everolimus intermediate supplier capable of meeting stringent global quality standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods, such as those documented in J. Labelled Compd. Radiopharm. (2000), rely on cumbersome reaction conditions that are ill-suited for large-scale industrial application. These conventional protocols typically employ a binary solvent system of toluene and dichloromethane (1:1 ratio) for the initial silylation step, requiring extended reaction times of up to 16 hours at moderate temperatures. The outcome of such processes is often disappointing, with reported yields for the key silylated intermediate hovering around a mere 6%. Furthermore, the subsequent deprotection step utilizes acetonitrile and demands rigorous temperature control, dropping to -45°C before warming to room temperature, yet still resulting in a final product yield of only 21%. These inefficiencies translate into excessive waste generation, high solvent consumption, and prohibitive raw material costs, rendering the process economically unfeasible for commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
In stark contrast, the methodology presented in Patent CN102127092B introduces a streamlined approach that dramatically improves process efficiency through solvent optimization and precise thermal management. The novel route utilizes pure toluene as the sole solvent for the silylation reaction, conducted at a tightly controlled range of 50-60°C. This adjustment not only accelerates the reaction kinetics, reducing the time to approximately 6 hours, but also boosts the isolated yield of the intermediate to 32%. Crucially, the process incorporates a strategic recovery mechanism where unreacted Rapamycin is reclaimed with a rate exceeding 50%, allowing for its reuse in subsequent batches. The second step employs tetrahydrofuran (THF) as the solvent for deprotection, initiating at 0°C to manage exothermicity before proceeding at room temperature, achieving a superior final yield of 66%. This represents a paradigm shift in cost reduction in pharmaceutical manufacturing by maximizing atom economy and minimizing waste.
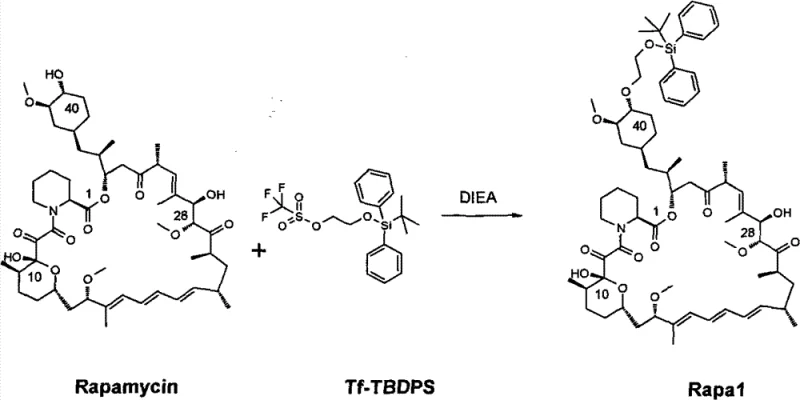
The visual representation of the first synthetic step highlights the selectivity achieved in protecting the 40-hydroxyl group of the Rapamycin macrocycle. The use of 2-(tert-butyldiphenylsilyl)ethoxytrifluoromethanesulfonate (Tf-TBDPS) as the silylating agent, in the presence of diisopropylethylamine (DIEA), ensures high regioselectivity. This specificity is vital for maintaining the biological activity of the final molecule, as incorrect protection at other hydroxyl sites (such as the 28-position) would render the API inactive. The structural integrity of the macrocyclic lactone and the triene system is preserved under these optimized toluene conditions, avoiding the degradation pathways often observed in harsher or prolonged reaction environments typical of older methodologies.
Mechanistic Insights into Silylation and Fluoride-Mediated Deprotection
The core of this synthetic strategy lies in the precise manipulation of protecting group chemistry to navigate the complex functionality of the Rapamycin scaffold. In the first stage, the mechanism involves the nucleophilic attack of the 40-hydroxyl oxygen on the silicon center of the Tf-TBDPS reagent. The triflate group serves as an excellent leaving group, facilitating the formation of the silyl ether bond. The choice of DIEA as a non-nucleophilic base is critical; it scavenges the triflic acid byproduct without interfering with the sensitive ketone or ester functionalities within the macrocycle. The reaction temperature of 50-60°C provides sufficient activation energy to overcome steric hindrance at the 40-position while remaining below the threshold where thermal decomposition of the starting material becomes significant. This balance is the key differentiator that allows for the higher yields observed in this patent compared to lower-temperature or mixed-solvent alternatives.
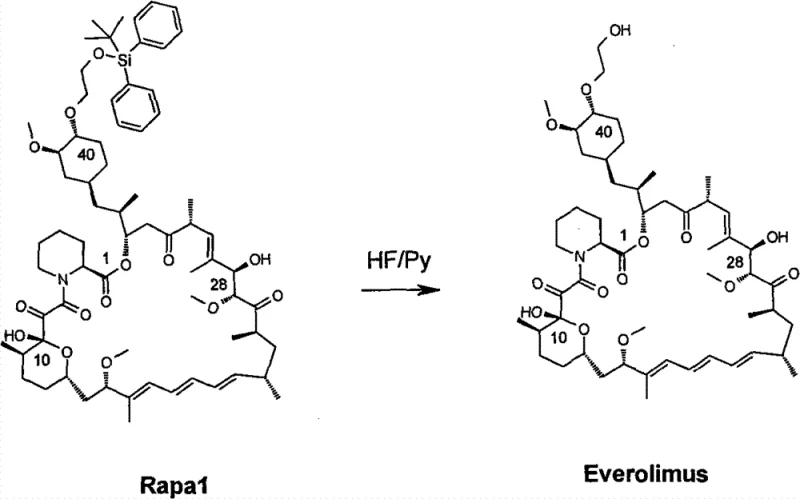
The second mechanistic phase involves the cleavage of the silyl ether to reveal the free hydroxyl group, completing the transformation to Everolimus. As depicted in the reaction scheme, the use of hydrogen fluoride-pyridine (HF/Py) complex in THF provides a source of nucleophilic fluoride ions. These ions have a high affinity for silicon, driving the formation of a strong Si-F bond and releasing the alcohol. The protocol specifies an initial temperature of 0°C because the formation of the Si-F bond is highly exothermic; uncontrolled heat release could lead to the isomerization of the sensitive conjugated triene system or hydrolysis of the lactone ring. By carefully managing this thermal profile and allowing the reaction to warm to room temperature overnight, the process ensures complete conversion while maintaining the stereochemical purity required for high-purity pharmaceutical intermediates. This meticulous control over reaction thermodynamics is essential for minimizing impurity profiles and simplifying downstream purification.
How to Synthesize Everolimus Efficiently
Implementing this synthesis requires strict adherence to the optimized parameters regarding solvent purity and temperature gradients to ensure reproducibility and safety. The process begins with the suspension of Rapamycin in anhydrous toluene, followed by the addition of the silylating agent and base under inert atmosphere to prevent moisture interference. Following the reaction, the workup involves a series of aqueous washes to remove salts and excess reagents, followed by concentration to a viscous oil. Purification is achieved via column chromatography using a petroleum ether and ethyl acetate gradient, which effectively separates the product from byproducts and allows for the distinct collection of unreacted starting material. The detailed standardized synthesis steps for this high-efficiency route are outlined below.
- React Rapamycin with 2-(tert-butyldiphenylsilyl)ethoxytrifluoromethanesulfonate and diisopropylethylamine in toluene at 50-60°C to form the silylated intermediate.
- Purify the intermediate via column chromatography and recover unreacted Rapamycin using ethyl acetate elution for recycling.
- Treat the purified intermediate with hydrogen fluoride-pyridine complex in tetrahydrofuran (THF), initiating at 0°C and warming to room temperature to obtain Everolimus.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology offers tangible benefits that extend beyond simple yield improvements. The ability to recover and recycle more than half of the expensive Rapamycin starting material fundamentally alters the cost structure of the production line. In traditional linear processes, unreacted starting material is often lost during workup or deemed too difficult to separate, representing a direct financial loss. By integrating a recovery loop, the effective consumption of the highest-cost input is drastically reduced, leading to substantial cost savings in the final API production. Furthermore, the simplification of the solvent system—moving from complex mixtures to single-solvent regimes like pure toluene and THF—streamlines solvent recovery and distillation operations, reducing both operational expenditure and environmental waste disposal costs.
- Cost Reduction in Manufacturing: The elimination of complex solvent mixtures and the reduction in reaction times directly correlate to lower utility and labor costs per kilogram of product. By avoiding the need for cryogenic cooling (-45°C) in the second step and utilizing milder conditions (0°C to RT), the energy demand for refrigeration is significantly curtailed. Additionally, the higher yields mean that less raw material is required to produce the same amount of finished goods, effectively lowering the variable cost of production without compromising on quality or purity specifications.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as toluene, THF, and ethyl acetate ensures that the supply chain is not vulnerable to shortages of exotic or highly regulated reagents. The robustness of the reaction conditions, particularly the tolerance for slightly broader temperature ranges in the first step (50-60°C), makes the process more forgiving and easier to control in a multi-purpose manufacturing plant. This reliability translates to more consistent batch cycles and reduced risk of production delays, ensuring a steady flow of high-purity pharmaceutical intermediates to downstream formulation partners.
- Scalability and Environmental Compliance: The process design inherently supports scalability by minimizing the number of unit operations and simplifying the purification workflow. While the patent describes column chromatography for isolation, the high selectivity of the reaction suggests that crystallization or extraction protocols could be developed for ton-scale production, further enhancing throughput. Moreover, the efficient use of solvents and the ability to recycle starting materials align with green chemistry principles, reducing the overall E-factor (mass of waste per mass of product) and facilitating compliance with increasingly stringent environmental regulations governing pharmaceutical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Everolimus synthesis route. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on process capabilities and limitations. Understanding these details is crucial for technical teams evaluating the feasibility of technology transfer or scale-up initiatives.
Q: What is the primary advantage of the toluene-based silylation method over conventional mixed-solvent systems?
A: The use of pure toluene at controlled temperatures (50-60°C) significantly simplifies the reaction matrix compared to traditional toluene/dichloromethane mixtures. This optimization reduces side reactions and degradation of the sensitive rapamycin macrocycle, leading to a marked increase in intermediate yield from approximately 6% in literature methods to over 30% in this patented process.
Q: How does the recovery of starting materials impact the overall cost structure?
A: Rapamycin is a high-value fermentation product. This process incorporates a specific recovery step where unreacted Rapamycin is eluted separately during purification. With recovery rates exceeding 50%, the effective consumption of the most expensive raw material is drastically reduced, directly lowering the cost of goods sold (COGS) for the final API intermediate.
Q: Why is the temperature control critical during the HF/Pyridine deprotection step?
A: The deprotection reaction is exothermic. Initiating the reaction at 0°C prevents thermal runaway which could degrade the thermolabile macrocyclic structure of the intermediate. Subsequent stirring at room temperature ensures complete conversion without compromising the structural integrity of the final Everolimus molecule.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Everolimus Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthetic routes in the competitive landscape of immunosuppressant manufacturing. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical advantages of patents like CN102127092B are fully realized in a GMP-compliant environment. We are committed to delivering high-purity Everolimus and its intermediates with stringent purity specifications, supported by our rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify identity and assay. Our infrastructure is designed to handle complex macrocyclic chemistry with the precision required for oncology and transplant medicines.
We invite potential partners to engage with our technical procurement team to discuss how our optimized manufacturing capabilities can support your supply chain goals. By leveraging our expertise in process optimization, we can provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us to request specific COA data and route feasibility assessments, ensuring that your project moves forward with a partner dedicated to quality, reliability, and continuous improvement in pharmaceutical intermediate supply.