Revolutionizing Cytidine Production: A Scalable Two-Step Chemical Synthesis for Global Pharma Markets
The pharmaceutical industry continuously seeks robust manufacturing pathways for critical nucleoside intermediates, and the technology disclosed in patent CN100488977C represents a significant leap forward in the chemical synthesis of cytidine. This specific intellectual property outlines a streamlined, two-step process that effectively bypasses the complex protection and deprotection sequences characteristic of legacy methodologies. By utilizing N4-acetylcytosine and beta-1,2,3,5-tetraacetyl ribofuranose as primary starting materials, the invention achieves a direct glycosylation followed by a mild alkaline deprotection. This approach not only simplifies the operational workflow but also ensures a high degree of stereochemical control, predominantly yielding the biologically active beta-configuration. For global procurement teams and R&D directors, understanding this shift from multi-step silylation routes to a direct acetylation strategy is crucial for evaluating long-term supply stability and cost structures in the antiviral and oncology sectors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the chemical synthesis of cytidine has been plagued by inefficient routes that rely heavily on expensive and difficult-to-handle reagents. For instance, early methods described by Nishimura involved the reaction of silyl ether-protected N4-acetylcytosine with 1-chlorotriphenylformyl ribose, a mixture of alpha and beta configurations that necessitated arduous separation processes to isolate the desired natural beta-cytidine. 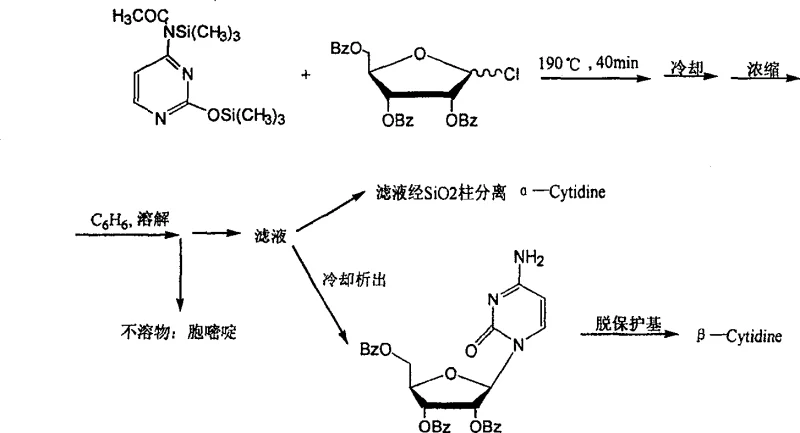 Furthermore, alternative pathways utilizing uridine as a starting material, such as those proposed by Helmut, required extreme reaction conditions including temperatures of 165°C and pressures of 25 atmospheres to effect ammonolysis.
Furthermore, alternative pathways utilizing uridine as a starting material, such as those proposed by Helmut, required extreme reaction conditions including temperatures of 165°C and pressures of 25 atmospheres to effect ammonolysis. 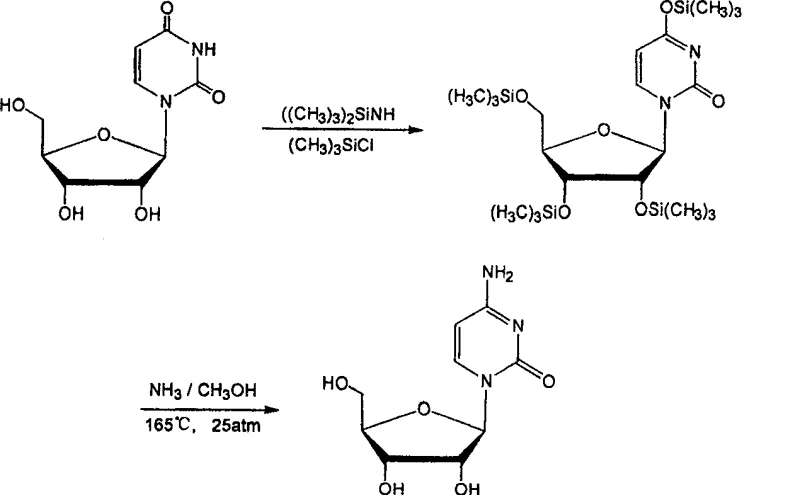 These conventional techniques suffer from low overall yields, complicated post-treatment procedures involving column chromatography, and the use of hazardous reagents that pose significant safety risks during commercial scale-up. The reliance on scarce biochemical reagents like uridine further exacerbates supply chain vulnerabilities, making these traditional methods economically unviable for large-volume API manufacturing.
These conventional techniques suffer from low overall yields, complicated post-treatment procedures involving column chromatography, and the use of hazardous reagents that pose significant safety risks during commercial scale-up. The reliance on scarce biochemical reagents like uridine further exacerbates supply chain vulnerabilities, making these traditional methods economically unviable for large-volume API manufacturing.
The Novel Approach
In stark contrast to the convoluted legacy processes, the novel approach detailed in the patent introduces a highly efficient condensation reaction catalyzed by phenolic phosphate compounds. This method allows for the direct coupling of N4-acetylcytosine with beta-1,2,3,5-tetraacetyl ribofuranose, either under solvent-free melting conditions or in the presence of organic solvents. 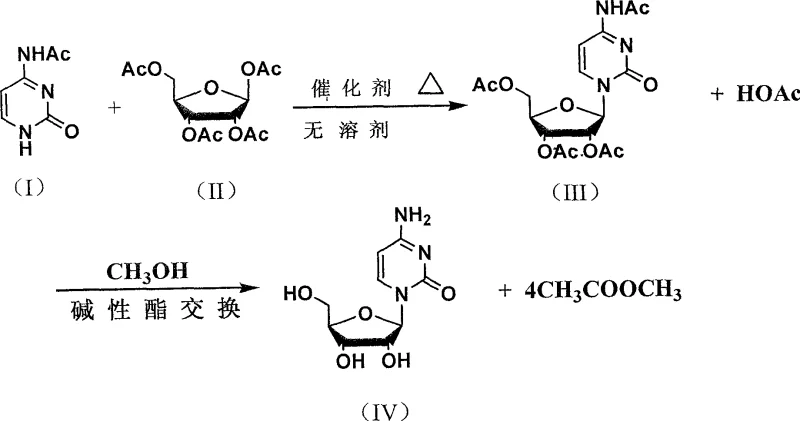 The innovation lies in the simultaneous removal of the acetic acid byproduct via vacuum distillation, which drives the equilibrium towards the formation of the intermediate N4-acetyl-1-(beta-D-2',3',5'-triacetyl ribofuranose)cytosine with conversion rates reaching 85-90%. Subsequent deprotection is achieved under mild conditions using common alkaline catalysts like sodium carbonate in methanol, avoiding the extreme pressures and temperatures of previous methods. This results in a total yield of 72-82%, a substantial improvement that translates directly into reduced waste generation and lower production costs for high-purity pharmaceutical intermediates.
The innovation lies in the simultaneous removal of the acetic acid byproduct via vacuum distillation, which drives the equilibrium towards the formation of the intermediate N4-acetyl-1-(beta-D-2',3',5'-triacetyl ribofuranose)cytosine with conversion rates reaching 85-90%. Subsequent deprotection is achieved under mild conditions using common alkaline catalysts like sodium carbonate in methanol, avoiding the extreme pressures and temperatures of previous methods. This results in a total yield of 72-82%, a substantial improvement that translates directly into reduced waste generation and lower production costs for high-purity pharmaceutical intermediates.
Mechanistic Insights into Phenolic Phosphate-Catalyzed Glycosylation
The core of this technological advancement rests on the specific catalytic activity of phenolic phosphate compounds, such as bis-p-nitrophenol phosphate or bis-p-fluorophenol phosphate, during the glycosylation step. These catalysts function effectively as Lewis acids, facilitating the formation of an oxocarbenium ion intermediate from the tetraacetyl ribofuranose. This activated sugar species is then attacked by the nitrogen atom at the N1 position of the N4-acetylcytosine base, establishing the critical N-glycosidic bond with high stereoselectivity for the beta-anomer. The use of a vacuum system to continuously remove the generated acetic acid is mechanistically vital, as it prevents the reverse hydrolysis reaction and pushes the equilibrium forward, ensuring high conversion efficiency without the need for excessive reagent loading. This precise control over the reaction environment minimizes the formation of alpha-isomers and other structural impurities, thereby simplifying the downstream purification requirements significantly.
Following the glycosylation, the deprotection mechanism involves a classic base-catalyzed ester exchange reaction, often referred to as transesterification or saponification depending on the specific base used. When the triacetyl intermediate is dissolved in methanol or ethanol with a catalytic amount of sodium carbonate or sodium hydroxide, the alkoxide ions attack the carbonyl carbons of the acetyl protecting groups. This cleavage releases acetate esters (such as methyl acetate) and regenerates the free hydroxyl groups on the ribose sugar, as well as the amino group on the cytosine base after spontaneous or facilitated deacetylation of the N4 position. The beauty of this mechanism is its specificity; the mild alkaline conditions are sufficient to cleave the ester bonds without degrading the sensitive glycosidic linkage or the pyrimidine ring, ensuring the structural integrity of the final cytidine product. This gentle deprotection stands in sharp contrast to the harsh ammonolysis required in older uridine-based routes, preserving product quality and reducing thermal stress on the manufacturing equipment.
How to Synthesize Cytidine Efficiently
The practical implementation of this synthesis route is designed for seamless integration into existing chemical manufacturing infrastructure, requiring standard stainless steel reactors equipped with heating, stirring, and vacuum capabilities. The process begins with the melting of the sugar derivative, followed by the controlled addition of the base and catalyst, allowing for a exothermic reaction to proceed safely under reflux. Detailed operational parameters, including specific temperature ramps and vacuum levels, are critical for maximizing the removal of acetic acid and preventing side reactions. For R&D teams looking to replicate or adapt this process, the following standardized synthesis steps outline the critical operational phases derived from the patent examples, ensuring reproducibility and safety during pilot and commercial runs.
- Heat beta-1,2,3,5-tetraacetyl ribofuranose to melt, then add N4-acetylcytosine and a phenolic phosphate catalyst under stirring to generate the protected intermediate while removing acetic acid byproduct via vacuum.
- Dissolve the intermediate N4-acetyl-1-(beta-D-2',3',5'-triacetyl ribofuranose)cytosine in methanol or ethanol, add an alkaline catalyst such as sodium carbonate, and reflux to remove acetyl groups.
- Purify the resulting crude cytidine through rectification to remove solvents and byproducts, followed by recrystallization from an ethanol-water mixture to obtain the final white columnar crystals.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, this patented methodology offers profound advantages by fundamentally altering the cost structure of cytidine production. The shift away from exotic starting materials like silylated uridine or chlorotriphenylmethyl ribose to commodity chemicals like tetraacetyl ribofuranose significantly lowers the barrier to entry and reduces raw material volatility. By eliminating the need for high-pressure autoclaves and extreme temperature conditions, the process reduces capital expenditure on specialized equipment and lowers energy consumption per kilogram of product. Furthermore, the simplified workup procedure, which relies on distillation and recrystallization rather than complex column chromatography, drastically reduces solvent usage and waste disposal costs. These factors combine to create a more resilient supply chain capable of meeting the growing demand for nucleoside analogs in the antiviral and oncology markets without the bottlenecks associated with older, less efficient technologies.
- Cost Reduction in Manufacturing: The elimination of expensive silylating agents and the use of inexpensive phenolic phosphate catalysts in minute quantities (0.5%-1.0%) leads to a substantial decrease in direct material costs. Additionally, the ability to operate under solvent-free conditions in the first step removes the cost of purchasing, recovering, and disposing of large volumes of organic solvents, further enhancing the economic viability of the process. The high conversion rates observed in the examples mean that less raw material is wasted as unreacted starting material or byproducts, optimizing the overall atom economy of the synthesis.
- Enhanced Supply Chain Reliability: By relying on widely available chemical feedstocks rather than scarce biochemical reagents, manufacturers can secure a more stable and continuous supply of raw materials, mitigating the risk of production stoppages due to shortages. The robustness of the reaction conditions, which do not require specialized high-pressure vessels, allows for production to be scaled across a broader range of facilities, increasing the overall capacity of the supply network. This flexibility ensures that procurement managers can maintain consistent inventory levels of high-purity pharmaceutical intermediates even during periods of market fluctuation.
- Scalability and Environmental Compliance: The process generates fewer hazardous byproducts and avoids the use of toxic heavy metal catalysts often found in alternative glycosylation methods, aligning with increasingly stringent environmental regulations. The straightforward isolation of the product through crystallization reduces the generation of liquid waste streams, simplifying effluent treatment and lowering the environmental footprint of the manufacturing site. This green chemistry profile not only ensures regulatory compliance but also enhances the corporate sustainability metrics for companies adopting this technology.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this cytidine synthesis technology. These answers are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on yield expectations, raw material specifications, and process safety. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their current manufacturing portfolios.
Q: How does this new synthesis method improve upon traditional silylation routes?
A: Unlike traditional methods requiring harsh ammonolysis conditions (165°C, 25atm) or expensive silylated reagents, this process utilizes mild glycosylation with phenolic phosphate catalysts, achieving total yields of 72-82% with significantly simpler post-processing.
Q: What are the primary cost drivers eliminated in this manufacturing process?
A: The process eliminates the need for costly starting materials like 1-chlorotriphenylformyl ribose or uridine derivatives, replacing them with readily available beta-1,2,3,5-tetraacetyl ribofuranose and N4-acetylcytosine, thereby drastically reducing raw material expenditure.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the method is designed for scalability, featuring a solvent-free option that minimizes waste and a straightforward deprotection step using common alkaline substances, ensuring consistent quality and high throughput for commercial supply chains.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cytidine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of optimized synthesis routes like the one described in CN100488977C for the global pharmaceutical supply chain. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent are fully realized in practical, GMP-compliant manufacturing environments. Our rigorous QC labs and stringent purity specifications guarantee that every batch of cytidine meets the exacting standards required for downstream drug development, providing our partners with the confidence needed to accelerate their clinical and commercial timelines.
We invite procurement leaders and R&D directors to engage with our technical procurement team to discuss how this advanced synthesis technology can be tailored to your specific volume requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the potential economic benefits of switching to this efficient route. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, ensuring that your supply of high-purity nucleoside intermediates remains secure, cost-effective, and aligned with the highest industry standards.