Advanced Synthesis of 5'-Deoxy-5-Fluorocytidine Derivatives for Safer Antineoplastic Therapy
The pharmaceutical landscape for antineoplastic agents is constantly evolving, driven by the urgent need to mitigate the severe side effects associated with traditional chemotherapy. Patent CN1022688C introduces a significant breakthrough in this domain through the development of novel 5'-deoxy-5-fluorocytidine derivatives. These compounds represent a sophisticated class of prodrugs designed to retain the potent antitumor activity of 5-fluorouracil while drastically reducing its notorious intestinal and immunosuppressive toxicity. For R&D directors and procurement specialists seeking reliable pharmaceutical intermediate suppliers, understanding the chemical architecture and synthetic feasibility of these derivatives is paramount. The patent details a versatile library of compounds wherein specific substituents at the N4, 2', and 3' positions are engineered to be readily hydrolyzable under physiological conditions, ensuring targeted drug release within the tumor microenvironment.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional administration of 5-fluorouracil (5-FU) and its immediate precursors, such as 5'-deoxy-5-fluorouridine, has long been hampered by a narrow therapeutic window. The primary bottleneck in conventional therapy is the non-selective toxicity that manifests as severe diarrhea and bone marrow suppression, often limiting the maximum tolerable dose before therapeutic efficacy is achieved. From a manufacturing perspective, the direct use of these active moieties often requires complex formulation strategies to mask their reactivity or control their release kinetics. Furthermore, the chemical instability of certain nucleoside analogues can lead to degradation during storage or transit, complicating the supply chain for high-purity API intermediates. The lack of selectivity means that healthy tissues are exposed to cytotoxic levels of the drug, leading to patient non-compliance and treatment discontinuation, which ultimately drives up the overall cost of care due to supportive treatments required for managing adverse events.
The Novel Approach
The innovation disclosed in CN1022688C circumvents these issues by modifying the nucleoside structure with physiologically hydrolyzable groups, effectively creating a prodrug system that is activated in vivo. By introducing acyl groups such as butyryl, benzoyl, or various substituted benzoyl moieties at the N4 position, the resulting derivatives exhibit significantly reduced acute toxicity while maintaining robust antitumor activity against sarcoma and carcinoma models. This structural modification not only enhances the lipophilicity and membrane permeability of the molecule but also alters its metabolic profile to favor tumor-selective activation. For supply chain heads, this approach offers a distinct advantage: the ability to synthesize a wide array of derivatives from a common core scaffold allows for rapid optimization of pharmacokinetic properties without necessitating entirely new synthetic routes. This flexibility supports cost reduction in API manufacturing by leveraging a unified platform technology for multiple potential drug candidates.
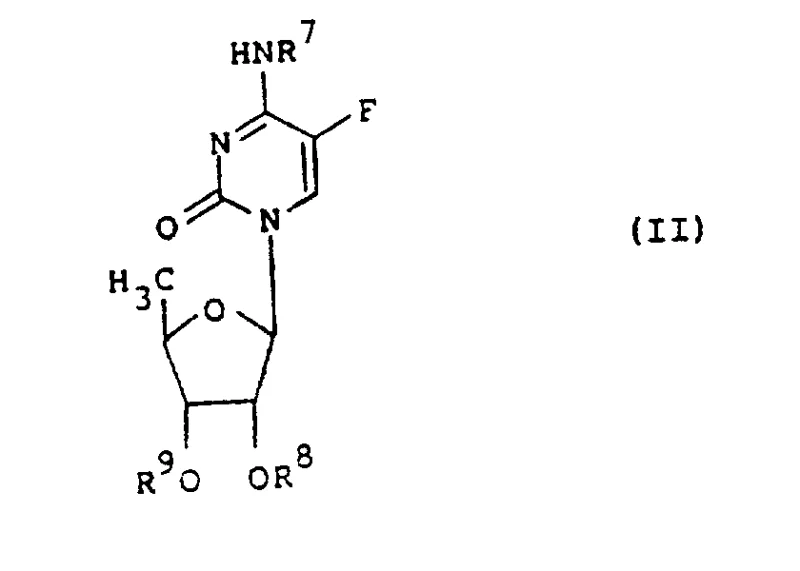
Mechanistic Insights into N4-Acylation and Prodrug Activation
The core chemical transformation involves the selective acylation of the exocyclic amine at the N4 position of the cytosine ring, often necessitating the temporary protection of the sugar hydroxyl groups to prevent O-acylation. The patent describes reacting a protected 5'-deoxy-5-fluorocytidine intermediate (Formula II) with activated acylating agents such as acid halides, acid anhydrides, or mixed anhydrides (Formula III). This nucleophilic acyl substitution is typically catalyzed by organic bases like pyridine or 4-dimethylaminopyridine (DMAP) in solvents such as dichloromethane or tetrahydrofuran. The choice of the R group is critical; it must be stable enough to survive the gastrointestinal tract yet labile enough to be cleaved by esterases or amidases once absorbed into the systemic circulation. This delicate balance ensures that the active 5-fluorouracil metabolite is released primarily at the site of action, thereby minimizing exposure to healthy gut mucosa.
Following the acylation step, the removal of protecting groups—such as tert-butyldimethylsilyl or isopropylidene groups—is performed under mild conditions to yield the final free hydroxyl derivatives. The mechanism of action relies on the intracellular conversion of these prodrugs into 5-fluoro-2'-deoxyuridine-5'-monophosphate (FdUMP), which inhibits thymidylate synthase, a key enzyme in DNA synthesis. By controlling the rate of hydrolysis through steric and electronic modulation of the N4 substituent, the pharmacokinetic profile can be tuned to sustain therapeutic plasma concentrations for longer durations. This mechanistic precision reduces the frequency of dosing required, which is a significant factor in improving patient compliance and overall treatment outcomes in clinical settings.
How to Synthesize 5'-Deoxy-5-Fluorocytidine Derivatives Efficiently
The synthesis of these high-value intermediates follows a logical progression of protection, functionalization, and deprotection, utilizing reagents that are commercially available and scalable. The process begins with the silylation of the ribose hydroxyls to direct the acylation exclusively to the N4 nitrogen, followed by reaction with the desired acid chloride or anhydride. Detailed standard operating procedures for scaling this reaction from gram to kilogram quantities involve strict temperature control, typically between 0°C and 50°C, to minimize side reactions and ensure high purity. For a comprehensive guide on the specific molar ratios, workup procedures, and purification methods such as silica gel chromatography or recrystallization, please refer to the standardized protocol below.
- Protect the hydroxyl groups of the starting 5'-deoxy-5-fluorocytidine using silyl protecting groups like tert-butyldimethylsilyl chloride in DMF.
- React the protected intermediate with an acylating agent (acid halide, anhydride, or mixed anhydride) in the presence of a base like pyridine or DMAP.
- Remove the protecting groups using fluoride sources or acidic hydrolysis to yield the final 5'-deoxy-5-fluorocytidine derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain strategists, the adoption of this synthetic route offers tangible benefits beyond mere chemical novelty. The versatility of the R group allows manufacturers to source a wide variety of carboxylic acid precursors, many of which are commodity chemicals available from multiple global vendors. This diversification of raw materials mitigates the risk of supply disruptions that often plague single-source API ingredients. Furthermore, the reaction conditions described in the patent avoid the use of exotic catalysts or extreme pressures, relying instead on standard batch reactor operations that are easily replicated in existing cGMP facilities. This compatibility with established infrastructure translates directly into substantial cost savings by eliminating the need for specialized equipment investments or extensive operator retraining programs.
- Cost Reduction in Manufacturing: The synthetic pathway utilizes widely available acylating agents and common organic solvents, which significantly lowers the raw material cost basis compared to more complex nucleoside modifications. By avoiding the need for transition metal catalysts, the downstream purification process is simplified, removing the expensive and time-consuming steps associated with heavy metal scavenging and residual analysis. This streamlined workflow enhances overall process efficiency, allowing for higher throughput and reduced cycle times, which ultimately drives down the cost per kilogram of the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The modular nature of the synthesis means that different derivatives can be produced on the same production line with minimal changeover time. This flexibility enables manufacturers to respond rapidly to fluctuating market demands for specific analogues without maintaining large inventories of finished goods. Additionally, the stability of the protected intermediates allows for strategic stockpiling at earlier stages of the synthesis, providing a buffer against potential logistical delays in the delivery of final reagents and ensuring continuous availability for downstream formulation partners.
- Scalability and Environmental Compliance: The processes outlined operate at moderate temperatures and atmospheric pressure, presenting fewer engineering challenges for scale-up from pilot plant to commercial production volumes. The waste streams generated are primarily composed of standard organic solvents and salts, which can be managed through conventional recovery and disposal systems, ensuring compliance with increasingly stringent environmental regulations. This robustness in process design facilitates the commercial scale-up of complex pharmaceutical intermediates while maintaining a sustainable manufacturing footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these 5'-deoxy-5-fluorocytidine derivatives. The answers are derived directly from the experimental data and claims presented in the patent literature, providing a factual basis for decision-making. Understanding these nuances is essential for evaluating the feasibility of integrating these intermediates into your current drug development pipeline or supply network.
Q: How do these derivatives improve upon traditional 5-Fluorouracil toxicity?
A: The derivatives described in CN1022688C act as prodrugs that are less toxic to the intestinal tract and immune system compared to 5-FU, offering a higher therapeutic index while maintaining antitumor efficacy.
Q: What represents the rate-limiting step in the manufacturing process?
A: The selective acylation at the N4 position without affecting the sugar moiety often requires precise protection-deprotection strategies, making the control of reaction temperature and stoichiometry critical.
Q: Are these intermediates scalable for commercial API production?
A: Yes, the patent outlines robust reaction conditions using common solvents like pyridine and dichloromethane, which are amenable to large-scale batch processing with standard purification techniques like recrystallization.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5'-Deoxy-5-Fluorocytidine Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of quality and consistency in the production of antineoplastic intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with unwavering reliability. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 5'-deoxy-5-fluorocytidine derivative meets the highest international standards for safety and efficacy. Our commitment to technical excellence allows us to navigate the complexities of nucleoside chemistry, delivering products that support the development of safer and more effective cancer therapies.
We invite you to collaborate with our technical procurement team to explore how our manufacturing capabilities can optimize your project timelines and budget. Contact us today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. Our experts are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced synthesis methods can enhance the value chain of your oncology portfolio.