Breaking Barriers in Local Anesthetic Purity: A New Era for Cinchocaine Hydrochloride Manufacturing
The pharmaceutical industry faces relentless pressure to deliver active pharmaceutical ingredients (APIs) and intermediates that meet increasingly stringent regulatory standards, particularly for potent local anesthetics like Cinchocaine Hydrochloride. Patent CN102633718B introduces a groundbreaking refinement methodology that addresses the critical limitations of prior art, specifically targeting the removal of stubborn structural impurities that have historically plagued manufacturing yields and purity profiles. This innovation is not merely a laboratory curiosity but a robust industrial solution designed to ensure compliance with United States Pharmacopeia (USP) regulations, which mandate that single impurities must not exceed 0.5% and total impurities must remain below 1.0%. By leveraging a sophisticated sequence of salt formation and recrystallization, this technology enables the production of Cinchocaine Hydrochloride with purity levels exceeding 99.5%, and in optimized embodiments, surpassing 99.9%. For global supply chains, this represents a pivotal shift towards more reliable pharmaceutical intermediates supplier capabilities, ensuring that downstream drug formulations are free from toxicological risks associated with residual byproducts.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Cinchocaine Hydrochloride has relied on methodologies documented in older patents such as US1825623, which typically involve reacting propyl carbinol with sodium metal followed by coupling with a quinoline derivative. While these methods establish the carbon skeleton of the molecule, the subsequent purification steps are notoriously inefficient at removing specific closely related byproducts. The conventional approach often utilizes ether precipitation to isolate the mono-hydrochloride salt, a technique that lacks the selectivity required to separate impurities with similar polarity and molecular weight. As illustrated in the structural analysis of potential byproducts, impurities labeled as 'a', 'b', 'c', and 'd' possess physicochemical properties that are nearly identical to the target molecule, making them exceptionally difficult to purge through simple washing or single-step crystallization. Consequently, products manufactured via these legacy routes frequently exhibit HPLC purities stagnating between 98.5% and 99.0%, with a persistent peak at a relative retention time (RRT) of approximately 0.9 that refuses to diminish regardless of repeated processing attempts.
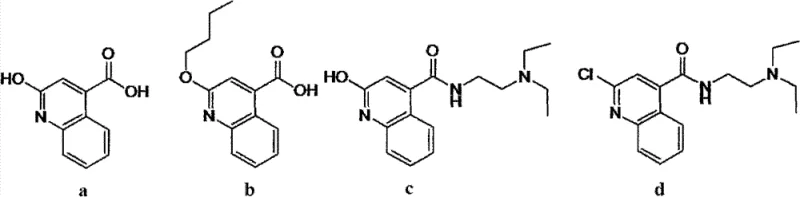
The Novel Approach
In stark contrast to the blunt instrument of ether precipitation, the novel approach detailed in CN102633718B employs a strategic "salt-switching" protocol that acts as a molecular filter. Instead of attempting to purify the final hydrochloride salt directly from the crude reaction mixture, the process first converts the crude base into a dihydrochloride salt in an acetone medium. This specific salt form exhibits differential solubility characteristics that allow the troublesome RRT 0.9 impurity to remain in the mother liquor while the desired product crystallizes out. Following this initial cleanup, the material is converted into an oxalate salt, a step that targets a different set of contaminants, specifically those with an RRT of roughly 0.75. This multi-stage purification strategy effectively deconstructs the impurity profile, tackling different contaminants at different stages of the synthesis rather than trying to remove them all at once. The result is a dramatic enhancement in product quality, transforming a marginally acceptable intermediate into a high-specification material suitable for sensitive medical applications.
Mechanistic Insights into Selective Salt Formation and Recrystallization
The core mechanism driving this purification success lies in the thermodynamic differences of solubility between the target molecule and its impurities when paired with different counter-ions. When crude Cinchocaine is treated with hydrochloric acid in acetone, it forms a dihydrochloride species. The crystal lattice energy of this di-salt is sufficiently distinct from that of the mono-protonated impurities, particularly the unreacted starting materials or hydrolysis products that contribute to the RRT 0.9 peak. By carefully controlling the temperature, typically cooling the reaction mixture to between 0°C and 5°C during acid addition and crystallization, the process maximizes the yield of the pure dihydrochloride while minimizing the occlusion of impurities. Subsequent recrystallization in ethanol further refines this intermediate, leveraging the temperature-dependent solubility curve of the dihydrochloride to exclude remaining trace contaminants. This step is critical because it sets the foundation for the final purity; if the RRT 0.9 impurity is not removed here, it is nearly impossible to eliminate in later stages due to its structural similarity to the final product.
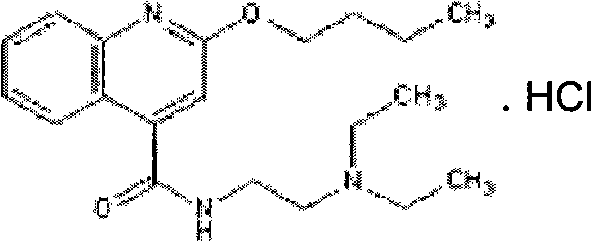
Following the dihydrochloride stage, the molecule is dissociated back to its free base form using an alkaline wash, typically with sodium hydroxide in a toluene system. This liquid-liquid extraction step serves a dual purpose: it regenerates the free amine and simultaneously washes away acidic or polar impurities that partition into the aqueous phase, effectively reducing the RRT 0.45 contaminant levels. The free base is then reacted with oxalic acid in absolute ethanol to form Cinchocaine Oxalate. The oxalate salt possesses a unique crystal habit and solubility profile that is highly effective at rejecting the RRT 0.75 impurity. By recrystallizing this oxalate salt multiple times in ethanol, the process achieves a level of purity where single impurities are driven down to below 0.1%. Finally, the ultra-pure oxalate is dissociated again and converted back to the hydrochloride salt in acetone. This final salification ensures the product is in its pharmacologically active and stable form, ready for formulation, with a melting point and spectral data consistent with the highest international standards.
How to Synthesize Cinchocaine Hydrochloride Efficiently
The synthesis of high-purity Cinchocaine Hydrochloride requires precise control over stoichiometry, temperature, and solvent ratios to ensure the selective crystallization mechanisms function as intended. The process begins with the dissolution of crude Cinchocaine in acetone, followed by the controlled addition of 36% hydrochloric acid at low temperatures to precipitate the dihydrochloride salt. After filtration and drying, this intermediate is recrystallized in ethanol to remove the bulk of the RRT 0.9 impurity. The purified dihydrochloride is then liberated to the free base using aqueous sodium hydroxide and extracted into toluene. This organic phase is reacted with oxalic acid dihydrate in ethanol to precipitate the oxalate salt, which undergoes further recrystallization to eliminate remaining trace byproducts. The final step involves liberating the base from the oxalate and re-salifying with hydrochloric acid in acetone to yield the final high-purity product. For detailed operational parameters, solvent ratios, and temperature profiles, please refer to the standardized synthesis guide below.
- React crude cinchocaine with hydrochloric acid in acetone to form cinchocaine dihydrochloride, followed by recrystallization in ethanol to remove RRT 0.9 impurities.
- Dissociate the dihydrochloride to free base, react with oxalic acid in absolute ethanol to form cinchocaine oxalate, and recrystallize to eliminate RRT 0.75 impurities.
- Alkalize the oxalate to free base in toluene, then react with hydrochloric acid in acetone to obtain high-purity cinchocaine hydrochloride with single impurities below 0.1%.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this advanced purification technology translates directly into enhanced operational efficiency and risk mitigation. Traditional methods often require extensive reprocessing or the use of expensive chromatographic techniques to meet purity specifications, which drives up manufacturing costs and extends lead times significantly. By implementing this salt-switching protocol, manufacturers can achieve compliance with USP standards through straightforward crystallization steps that utilize commodity solvents like acetone, ethanol, and toluene. This eliminates the need for specialized separation equipment or exotic reagents, thereby simplifying the supply chain and reducing the dependency on single-source specialty chemical vendors. Furthermore, the robustness of the process ensures consistent batch-to-batch quality, reducing the likelihood of costly batch failures or regulatory rejection during quality control audits.
- Cost Reduction in Manufacturing: The elimination of complex purification steps such as preparative HPLC or multiple ether washes results in substantial cost savings. By relying on crystallization-driven purification, the process minimizes solvent consumption and waste generation, leading to a more environmentally friendly and economically viable production model. The use of common solvents also allows for efficient recovery and recycling systems, further driving down the variable costs associated with raw material consumption.
- Enhanced Supply Chain Reliability: Because the process does not rely on fragile catalytic systems or unstable intermediates, it offers superior scalability and reproducibility. This stability is crucial for maintaining continuous supply lines to downstream pharmaceutical formulators who cannot afford interruptions in their API sourcing. The ability to consistently produce material with single impurities below 0.1% ensures that customers receive a product that is ready for immediate use in final drug formulations without additional incoming quality testing delays.
- Scalability and Environmental Compliance: The methodology is inherently scalable, having been validated from laboratory bench scales up to multi-kilogram pilot batches with consistent results. The waste streams generated are primarily composed of standard organic solvents and aqueous salt solutions, which are well-understood and easily managed within modern wastewater treatment facilities. This aligns with global trends towards greener chemistry and helps manufacturing partners meet increasingly strict environmental, social, and governance (ESG) targets without compromising on product quality or output volume.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and benefits of this purification technology. Understanding these details is essential for R&D teams evaluating the feasibility of integrating this process into existing manufacturing lines. The answers provided are derived directly from the experimental data and technical disclosures found in the patent literature, ensuring accuracy and relevance for technical decision-makers.
Q: Why do conventional methods fail to meet USP standards for Cinchocaine?
A: Conventional methods, such as those described in US1825623, often result in products with purity around 98.5-99.0%. They struggle to remove structurally similar impurities, particularly those with a relative retention time (RRT) of roughly 0.9, which co-crystallize with the product during standard ether precipitation.
Q: How does the salt-switching technique improve impurity profiles?
A: By sequentially converting the molecule into a dihydrochloride salt and then an oxalate salt, the process exploits subtle differences in solubility between the API and its byproducts. This selective crystallization effectively strips away specific contaminants like RRT 0.45, 0.75, and 0.9 that are otherwise inseparable.
Q: Is this purification method suitable for large-scale commercial production?
A: Yes, the process utilizes common, industrially available solvents such as acetone, ethanol, and toluene. It avoids exotic reagents or complex chromatographic separations, making it highly amenable to commercial scale-up while maintaining stringent quality control standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cinchocaine Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and a commitment to quality excellence. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated purification techniques described in CN102633718B can be seamlessly integrated into your supply chain. We maintain stringent purity specifications across all our product lines, supported by rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify that every batch meets or exceeds the 99.9% purity benchmark. Whether you require custom synthesis of complex intermediates or large-scale supply of established anesthetics, our infrastructure is designed to support your growth and regulatory compliance needs.
We invite you to engage with our technical procurement team to discuss how we can optimize your current sourcing strategy. By requesting a Customized Cost-Saving Analysis, you can gain valuable insights into how switching to our advanced purification grades can reduce your overall cost of goods sold. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments tailored to your specific project requirements. Let us help you secure a supply of high-purity Cinchocaine Hydrochloride that empowers your pharmaceutical development pipeline with confidence and reliability.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →