Advanced Synthesis of Targeted Ursolic Acid Derivatives for Commercial Scale-Up
Advanced Synthesis of Targeted Ursolic Acid Derivatives for Commercial Scale-Up
The pharmaceutical industry is constantly seeking novel scaffolds that offer improved therapeutic indices over natural products, and Patent CN102516351A presents a significant breakthrough in the semi-synthesis of ursolic acid derivatives. This intellectual property details a robust methodology for modifying the pentacyclic triterpenoid skeleton of ursolic acid to create compounds with superior anti-proliferative activity against various tumor cell lines including HepG2, BGC823, and Hela. Unlike traditional extraction methods which yield the parent compound with limited solubility and moderate potency, this patented approach utilizes precise chemical modifications at the C-3 and C-28 positions to enhance bioavailability and introduce tumor-targeting capabilities. For R&D directors and procurement specialists, understanding this synthetic pathway is crucial as it represents a viable route for producing high-purity pharmaceutical intermediates that address the critical need for effective anti-cancer agents with reduced systemic toxicity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the utilization of ursolic acid in drug development has been hindered by its poor water solubility and lack of specific targeting mechanisms, which often results in high dosage requirements and potential off-target effects. Conventional isolation from plant sources yields the native structure, which, while biologically active, suffers from pharmacokinetic limitations that restrict its clinical efficacy in treating aggressive malignancies. Furthermore, direct modification of the native molecule without protecting group strategies often leads to complex mixtures of regioisomers, complicating downstream purification and significantly driving up manufacturing costs. The absence of a defined linker system in native ursolic acid also prevents the attachment of targeting moieties, such as folate, which are essential for modern precision medicine approaches aimed at minimizing damage to healthy tissues like human embryonic lung fibroblasts.
The Novel Approach
The methodology outlined in the patent overcomes these hurdles through a strategic three-step synthetic sequence that ensures high regioselectivity and structural integrity of the final products. By initially acetylating the 3-hydroxyl group, the process not only protects this sensitive site but also modulates the lipophilic character of the molecule, which is critical for membrane permeability. Subsequent activation of the C-28 carboxylic acid allows for the introduction of an ethylenediamine linker, creating a versatile handle for further functionalization with targeting ligands like folic acid. This modular approach enables the production of distinct derivatives, such as the folate-conjugated Compound I and the hydroxy-containing Compound IV, each tailored for specific biological interactions while maintaining a scalable and reproducible manufacturing profile suitable for industrial adoption.
Mechanistic Insights into Multi-Step Functionalization of Triterpenoids
The core of this synthetic strategy relies on classic organic transformations optimized for the sterically crowded environment of the ursane skeleton. The initial esterification involves the nucleophilic attack of the 3-beta hydroxyl group on acetic anhydride, catalyzed by DMAP in pyridine, which proceeds efficiently at room temperature to yield the 3-O-acetyl intermediate. Following this, the carboxylic acid at position 28 is activated using oxalyl chloride to form a reactive acyl chloride species, which is immediately subjected to nucleophilic substitution by ethylenediamine. This amidation step is critical as it installs the primary amine functionality required for the final conjugation, and the use of triethylamine as a base ensures the neutralization of generated HCl, driving the reaction to completion without degrading the sensitive triterpenoid core.
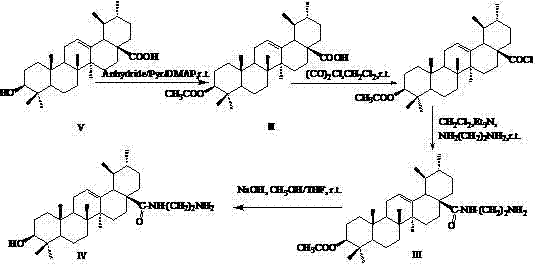
For the targeted derivative, the final coupling reaction utilizes carbodiimide chemistry, specifically EDC and NHS, to activate the carboxylic acid of folic acid for amide bond formation with the diamine linker. This bioconjugation step must be performed under anhydrous conditions in DMSO to prevent hydrolysis of the activated ester, ensuring high coupling efficiency. Alternatively, for non-targeted variants, a simple hydrolysis step using sodium hydroxide removes the acetyl protecting group to reveal the free 3-hydroxyl moiety, demonstrating the flexibility of this platform to generate a library of analogs. The rigorous purification protocols, involving column chromatography and recrystallization, ensure that the final intermediates meet the stringent purity specifications required for preclinical and clinical evaluation.
How to Synthesize N-[3β-acetoxy-arbutane-12-en-28-yl]-aminoethylenediamine Efficiently
The preparation of these high-value intermediates requires strict adherence to the reaction parameters defined in the patent to maximize yield and minimize impurity profiles. The process begins with the dissolution of the starting material in appropriate solvents, followed by the controlled addition of reagents to manage exothermic events and ensure homogeneous reaction conditions. Detailed operational guidelines regarding pH adjustments, solvent removal, and crystallization temperatures are essential for operators to replicate the laboratory success on a larger pilot or production scale. The standardized synthetic steps provided below outline the critical control points necessary for achieving consistent quality in the manufacturing of these complex triterpenoid derivatives.
- Acetylate ursolic acid using acetic anhydride and DMAP in pyridine to protect the 3-hydroxyl group and form 3-O-acetyl ursolic acid.
- Activate the carboxylic acid at position 28 with oxalyl chloride, followed by reaction with ethylenediamine to form the amide intermediate.
- Conjugate the amine intermediate with folic acid using EDC/NHS coupling agents, or hydrolyze the acetyl group to obtain the hydroxy derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, this synthetic route offers substantial advantages due to its reliance on readily available and cost-effective reagents that are standard in the fine chemical industry. The use of common solvents like dichloromethane, pyridine, and ethanol simplifies the procurement process and reduces the risk of supply disruptions associated with exotic or highly regulated chemicals. Furthermore, the reaction conditions are predominantly conducted at room temperature, which significantly lowers energy consumption compared to processes requiring cryogenic cooling or high-temperature reflux, thereby contributing to overall cost reduction in API manufacturing. The ability to purify intermediates using standard silica gel chromatography and recrystallization techniques also means that existing infrastructure can be utilized without the need for specialized equipment investments.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the use of stoichiometric reagents like oxalyl chloride and acetic anhydride allow for predictable cost modeling and significant savings on raw materials. By avoiding complex catalytic cycles that require rigorous removal of trace metals, the downstream processing is simplified, reducing the burden on quality control laboratories and shortening the batch cycle time. This streamlined approach ensures that the cost of goods sold remains competitive, making these derivatives attractive candidates for further development in cost-sensitive markets.
- Enhanced Supply Chain Reliability: Since the starting material, ursolic acid, is a natural product widely distributed in plants, the supply base is robust and less susceptible to the geopolitical volatility often seen with synthetic petrochemical feedstocks. The synthetic steps are robust and tolerant to minor variations in reaction parameters, which enhances the reliability of supply by reducing the rate of batch failures. This stability allows for long-term planning and secure contracting, ensuring that pharmaceutical partners can maintain continuous production schedules for their final drug formulations.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing unit operations that translate easily from gram-scale laboratory synthesis to multi-kilogram production runs. The waste streams generated, primarily consisting of aqueous acidic washes and organic solvents, can be managed through standard treatment protocols, facilitating compliance with environmental regulations. The high atom economy of the amidation and esterification steps further minimizes waste generation, aligning with green chemistry principles and supporting the sustainability goals of modern pharmaceutical manufacturers.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the synthesis and application of these ursolic acid derivatives, based on the detailed experimental data provided in the patent documentation. Understanding these aspects is vital for technical teams evaluating the feasibility of integrating these intermediates into their drug discovery pipelines. The answers reflect the specific chemical behaviors and biological outcomes observed during the development of this technology.
Q: How does the folate conjugation improve the therapeutic index of ursolic acid derivatives?
A: The conjugation with folic acid targets folate receptors which are often overexpressed on tumor cells, thereby increasing drug accumulation in cancer tissue while reducing toxicity to normal cells like HELF fibroblasts.
Q: What are the key purification methods described for these intermediates?
A: The patent describes column chromatography using petroleum ether/ethyl acetate systems for intermediates and reverse-phase column chromatography with methanol/water for the final folate-conjugated product, followed by recrystallization.
Q: Why is the 3-position acetylation significant in this synthesis route?
A: Acetylation at the 3-beta position modifies the lipophilicity and metabolic stability of the triterpenoid skeleton, contributing to the observed enhancement in cytotoxicity against cell lines like HepG2 and Hela compared to native ursolic acid.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ursolic Acid Derivative Supplier
As a leading CDMO partner, NINGBO INNO PHARMCHEM possesses the technical expertise and infrastructure to scale these complex synthetic pathways from 100 kgs to 100 MT/annual commercial production with exceptional precision. Our facilities are equipped with state-of-the-art reactors and purification systems capable of handling the specific solvent systems and reaction conditions required for triterpenoid modification, ensuring stringent purity specifications are met for every batch. We understand the critical nature of supply continuity in the pharmaceutical sector and have established rigorous QC labs to monitor every step of the synthesis, guaranteeing that our clients receive intermediates that are fully compliant with global regulatory standards.
We invite potential partners to engage with our technical procurement team to discuss how we can support your specific project requirements through a Customized Cost-Saving Analysis. By leveraging our process optimization capabilities, we can help you navigate the complexities of scaling this chemistry, offering specific COA data and route feasibility assessments tailored to your timeline. Contact us today to explore how our reliable ursolic acid derivative supplier services can accelerate your path to clinical success.