Advanced Valsartan Refining Technology: Eliminating Nitrosamines for Global Pharmaceutical Supply Chains
Advanced Valsartan Refining Technology: Eliminating Nitrosamines for Global Pharmaceutical Supply Chains
The pharmaceutical industry faces unprecedented scrutiny regarding genotoxic impurities, particularly nitrosamines in angiotensin II receptor blockers. Patent CN113993851A introduces a groundbreaking refining method specifically designed to address the critical challenge of N-nitrosodimethylamine (NDMA) contamination in Valsartan. This technology leverages a sophisticated acid-base salt formation strategy followed by selective crystallization to achieve purity levels that exceed current regulatory standards. By transforming the crude drug substance into a water-soluble salt and subsequently regenerating the free acid, the process effectively partitions lipophilic nitrosamine impurities away from the active pharmaceutical ingredient. This approach not only ensures patient safety by reducing NDMA levels to ≤0.03ppm but also offers a robust, scalable pathway for manufacturers seeking to stabilize their supply chains against regulatory disruptions.
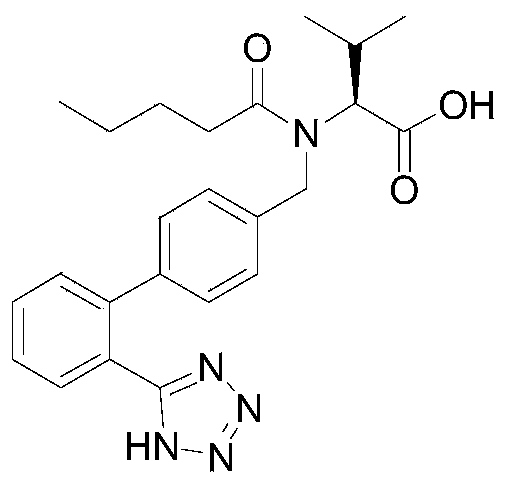
Valsartan, chemically known as N-(1-valeryl)-N-[[2'-(1H-tetrazol-5-yl)[1,1'-biphenyl]-4-yl]methyl]-L-valine, is a cornerstone medication for hypertension management. As depicted in the structural analysis, the molecule contains a complex biphenyl tetrazole framework that presents unique purification challenges. The presence of the tetrazole ring, essential for its biological activity as an AT1 receptor antagonist, often necessitates synthetic routes involving azides and high-boiling solvents. These historical synthetic pathways have inadvertently introduced nitrosamine risks, creating an urgent demand for downstream processing technologies that can remediate these impurities without compromising the structural integrity or yield of the final API. The method described in CN113993851A directly targets these pain points by decoupling the purification logic from the synthesis constraints.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional purification strategies for Valsartan have predominantly relied on simple recrystallization from organic solvents such as ethyl acetate, isopropyl ether, or mixed solvent systems. While these methods can improve chemical purity to some extent, they often fail to adequately remove trace nitrosamine contaminants due to similar solubility profiles between the impurity and the drug substance. Furthermore, repeated recrystallization cycles frequently lead to significant yield losses, sometimes rendering the process economically unviable for large-scale production. Another critical drawback is the potential for polymorphic transformation; aggressive solvent changes or excessive thermal cycling during conventional work-ups can induce unwanted crystal form changes, which may alter the bioavailability and stability of the final drug product. These limitations underscore the necessity for a more discriminating purification technique that operates on fundamental physicochemical differences rather than simple solubility gradients.
The Novel Approach
The innovative methodology disclosed in the patent circumvents these issues by exploiting the acidic nature of the tetrazole ring in Valsartan. Instead of direct recrystallization, the process converts the crude Valsartan into a water-soluble salt using alkalis like sodium hydroxide or weak acid salts like sodium carbonate. This phase transfer allows for the separation of water-insoluble organic impurities, including many nitrosamine precursors, in the aqueous phase. Subsequent acidification regenerates the free acid form of Valsartan as a precipitate, leaving water-soluble inorganic salts and polar impurities behind. By repeating this acid-base cycle, the purity is incrementally enhanced without the need for exotic chromatography or hazardous solvent exchanges. The final step involves a controlled crystallization from ethyl acetate, which polishes the product to pharmaceutical grade specifications while maintaining a stable crystal lattice structure suitable for formulation.
Mechanistic Insights into Nitrosamine Formation and Removal
Understanding the origin of NDMA is crucial for appreciating the efficacy of this refining method. In many Valsartan synthesis routes, the formation of the tetrazole ring requires the use of sodium azide in polar aprotic solvents like N,N-dimethylformamide (DMF). Under the acidic conditions often present during work-up or quenching with sodium nitrite, DMF can hydrolyze to release dimethylamine. This secondary amine then reacts with nitrosating agents to form NDMA, a potent carcinogen. The reaction mechanism illustrates how the decomposition of the solvent itself becomes the source of the contaminant, creating a persistent risk that is difficult to eliminate through standard washing procedures alone. This intrinsic link between the synthetic solvent and the impurity profile demands a purification strategy that does not rely on the same solvent systems used in the upstream synthesis.
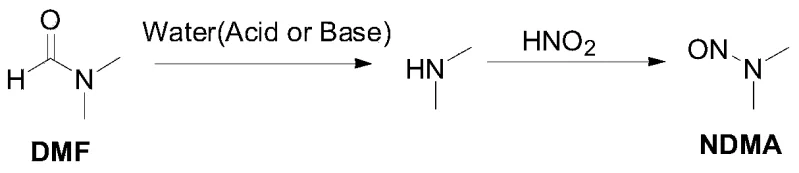
The refining process described in CN113993851A effectively breaks this contamination cycle by shifting the purification medium to water and ethyl acetate. Since the acid-base salt formation occurs in an aqueous environment, the partition coefficient of NDMA favors the organic phase or allows it to remain in the mother liquor upon precipitation of the Valsartan salt. When the Valsartan is regenerated via acidification, the NDMA, being highly soluble in organic phases and relatively volatile, is largely excluded from the precipitating solid. The final dissolution in ethyl acetate serves as a polishing step where any residual traces are kept in solution while the pure Valsartan crystallizes out upon cooling. This multi-stage orthogonal purification ensures that the thermodynamic driving force favors the exclusion of the nitrosamine impurity at every step, resulting in a final product with NDMA levels consistently below the strict 0.03ppm threshold mandated by global health authorities.
How to Synthesize High-Purity Valsartan Efficiently
Implementing this refining protocol requires precise control over pH and temperature to maximize yield and purity. The process begins with the dispersion of crude Valsartan in water, followed by the careful addition of a base to adjust the pH to a range of 7 to 12, ensuring complete salt formation without degrading the sensitive tetrazole ring. After filtering to remove insolubles, the solution is acidified to a pH of 0.5 to 2.0 to precipitate the purified drug. This cycle may be repeated multiple times depending on the initial impurity load. The final crystallization from ethyl acetate must be managed with controlled cooling rates to ensure optimal crystal growth and filtration characteristics. For a detailed breakdown of the specific reagent quantities, reaction times, and temperature profiles validated in the patent examples, please refer to the standardized synthesis guide below.
- Dissolve crude Valsartan in water and react with an alkali or strong base weak acid salt to form a Valsartan salt solution.
- Acidify the salt solution with an inorganic or organic acid to regenerate Valsartan precipitate, then filter to obtain a purified crude product.
- Repeat the salt formation and acidification steps to further reduce impurities, then dissolve the final crude product in ethyl acetate for crystallization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this refining technology translates into tangible strategic benefits beyond mere regulatory compliance. The shift towards an aqueous-based purification system significantly reduces the dependency on expensive, high-boiling organic solvents that are costly to recover and dispose of. By simplifying the unit operations to basic salt formation and precipitation, the process becomes inherently more robust and easier to scale from pilot batches to multi-ton commercial production without the need for specialized high-pressure equipment. This operational simplicity directly correlates to reduced capital expenditure and lower operating costs, making the supply of high-purity Valsartan more resilient against market fluctuations in raw material prices. Furthermore, the ability to consistently meet the stringent 0.03ppm NDMA limit mitigates the risk of costly product recalls and market withdrawals, safeguarding the long-term viability of the supply contract.
- Cost Reduction in Manufacturing: The elimination of complex chromatographic purification steps and the reduction in organic solvent usage lead to substantial cost savings. By utilizing water as the primary medium for the bulk of the purification, the energy load associated with solvent distillation and recovery is drastically minimized. Additionally, the high yields reported (80-86%) mean that less starting material is wasted, optimizing the overall material balance and reducing the cost of goods sold (COGS) for the final API.
- Enhanced Supply Chain Reliability: The use of commodity chemicals such as sodium hydroxide, hydrochloric acid, and ethyl acetate ensures that the supply chain is not vulnerable to shortages of exotic reagents. The robustness of the acid-base extraction method allows for consistent batch-to-batch quality, reducing the variability that often plagues complex synthetic processes. This reliability enables procurement teams to forecast inventory needs with greater accuracy and maintain leaner stock levels without compromising on delivery schedules.
- Scalability and Environmental Compliance: From an environmental perspective, the reduction in hazardous waste generation is significant. The aqueous waste streams generated during the salt formation steps are easier to treat compared to mixed organic waste, facilitating compliance with increasingly strict environmental regulations. The process is designed for industrial production, meaning it can be seamlessly integrated into existing manufacturing facilities with minimal retrofitting, allowing for rapid scale-up to meet surging global demand for safe antihypertensive medications.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Valsartan refining technology. These insights are derived directly from the experimental data and technical disclosures within patent CN113993851A, providing a clear understanding of the process capabilities and limitations. Understanding these details is essential for technical teams evaluating the feasibility of integrating this method into their current manufacturing workflows.
Q: What is the acceptable limit for NDMA in Valsartan according to this patent?
A: The patent describes a method capable of reducing nitrosamine impurities (NDMA) to less than or equal to 0.03ppm (30 ppb), meeting the stringent requirements set by the European Pharmacopoeia Commission (EDQM).
Q: Does this refining process use hazardous organic solvents?
A: The core purification steps utilize water as the primary solvent for the acid-base conversion. Ethyl acetate is used only in the final crystallization step, significantly reducing the reliance on high-boiling point aprotic solvents like DMF during the purification phase.
Q: What yields can be expected from this refining method?
A: Experimental data in the patent indicates that the method achieves high yields ranging from 80% to 86%, while simultaneously obtaining HPLC purity of 99.9%.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Valsartan Supplier
At NINGBO INNO PHARMCHEM, we recognize that the purity of Valsartan is not just a specification but a commitment to patient safety. Our technical team has extensively analyzed advanced refining protocols like the one described in CN113993851A to ensure our manufacturing processes are at the forefront of impurity control. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab-scale optimization to full-scale manufacturing is seamless. Our facilities are equipped with rigorous QC labs and stringent purity specifications that go beyond standard pharmacopoeial requirements, guaranteeing that every batch of Valsartan we supply meets the lowest possible limits for genotoxic impurities.
We invite global pharmaceutical partners to collaborate with us on securing a stable supply of high-quality Valsartan. Our team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team to request specific COA data, route feasibility assessments, and samples for your validation studies. Let us help you navigate the complexities of nitrosamine control and secure your supply chain with a partner dedicated to excellence and compliance.