Advanced Purification Strategy for High-Purity 16α-Hydroxyprednisolone Intermediates
The pharmaceutical industry continuously demands higher purity standards for corticosteroid intermediates, particularly for potent anti-inflammatory agents like 16α-hydroxyprednisolone. Patent CN112125943A introduces a groundbreaking preparation method that addresses the persistent challenge of removing structurally similar byproducts, specifically designated as Impurity H, which often plague traditional synthesis routes starting from tetraene acetate derivatives. This technical disclosure outlines a sophisticated yet operationally simple strategy involving selective oxidation followed by precise hydrolysis, enabling manufacturers to achieve purity levels exceeding 99.5% with impurity content suppressed below 0.02%. For R&D directors and process chemists, this represents a significant leap forward in controlling the杂质谱 (impurity profile) of complex steroid backbones without resorting to expensive chromatographic separations. The methodology leverages common reagents like hypochlorites and organic acids under mild thermal conditions, suggesting a robust pathway that is highly amenable to large-scale industrial adaptation while maintaining rigorous quality control standards required for downstream API synthesis.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for 16α-hydroxyprednisolone, particularly those utilizing tetraene acetate as a starting material, suffer from a critical bottleneck: the formation of Impurity H. As illustrated in the reaction scheme below, this impurity arises during the multi-step transformation involving bromination and hydroxylation, resulting in a molecular structure that is nearly identical to the desired product. 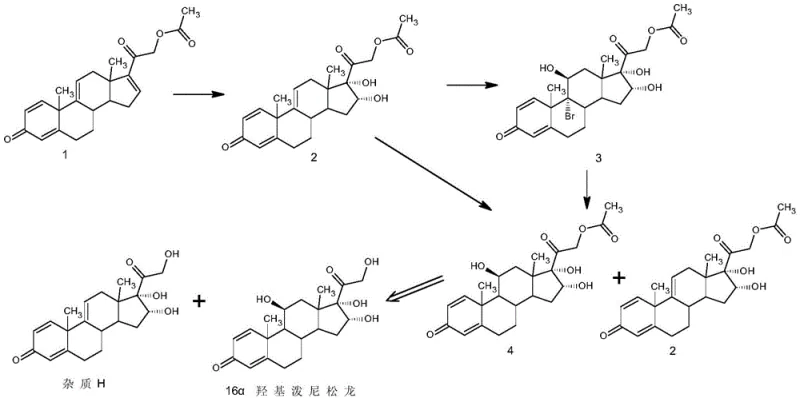 Because Impurity H shares such close structural homology with 16α-hydroxyprednisolone, their physical properties, such as solubility and melting point, are remarkably similar. Consequently, standard purification techniques like simple recrystallization or washing are largely ineffective, often leaving residual impurity levels around 0.90% or higher. This level of contamination is unacceptable for high-grade pharmaceutical applications, necessitating complex and costly additional purification steps that drastically reduce overall yield and increase production lead times. The inability to efficiently separate these congeners has historically restricted the widespread industrial adoption of otherwise cost-effective synthetic routes based on tetraene acetate precursors.
Because Impurity H shares such close structural homology with 16α-hydroxyprednisolone, their physical properties, such as solubility and melting point, are remarkably similar. Consequently, standard purification techniques like simple recrystallization or washing are largely ineffective, often leaving residual impurity levels around 0.90% or higher. This level of contamination is unacceptable for high-grade pharmaceutical applications, necessitating complex and costly additional purification steps that drastically reduce overall yield and increase production lead times. The inability to efficiently separate these congeners has historically restricted the widespread industrial adoption of otherwise cost-effective synthetic routes based on tetraene acetate precursors.
The Novel Approach
The innovative process described in the patent circumvents these separation difficulties by chemically modifying the impurity rather than attempting physical separation alone. By treating the crude 16α-hydroxyprednisolone acetate with an aqueous hypochlorite solution in the presence of an organic acid, the process selectively targets the reactive sites present in Impurity H. This oxidative treatment converts the stubborn impurity into more polar or soluble derivatives that no longer co-crystallize with the product. Following this reaction, a water precipitation step effectively isolates the purified acetate intermediate, leaving the modified impurities in the aqueous mother liquor. This strategic intervention transforms a difficult physical separation problem into a manageable chemical conversion, ensuring that the subsequent hydrolysis step begins with a significantly cleaner substrate. The result is a streamlined workflow that delivers high-purity material suitable for direct use in final API manufacturing without the need for extensive downstream processing.
Mechanistic Insights into Hypochlorite-Mediated Impurity Removal
The core of this technological advancement lies in the selective reactivity of hypochlorite species towards the specific functional groups distinguishing Impurity H from the target molecule. While the exact mechanistic details involve complex steroid chemistry, the process relies on the differential oxidation potential between the desired 16α-hydroxyprednisolone acetate and its byproduct. Under controlled temperatures ranging from 15°C to 40°C, the hypochlorite ion acts as a mild oxidizing agent that likely targets electron-rich double bonds or specific hydroxyl configurations unique to the impurity structure. The addition of organic acids such as formic acid or acetic acid helps modulate the pH and reactivity of the hypochlorite, preventing over-oxidation of the valuable product while ensuring complete conversion of the contaminant. This fine-tuned reaction environment is critical; too aggressive conditions could degrade the steroid backbone, while too mild conditions would fail to eliminate the impurity. The patent data confirms that maintaining this balance allows for the preservation of the stereochemical integrity at the 16α position, which is vital for the biological activity of the final drug substance.
Following the oxidative treatment, the process employs a quenching step using sodium bisulfite to neutralize excess oxidants, preventing any further unwanted reactions during workup. The subsequent concentration and water precipitation leverage the changed solubility profile of the oxidized impurity. Since the modified impurity is now more hydrophilic or structurally disrupted, it remains dissolved in the aqueous phase or forms oils that do not incorporate into the crystal lattice of the precipitating acetate. 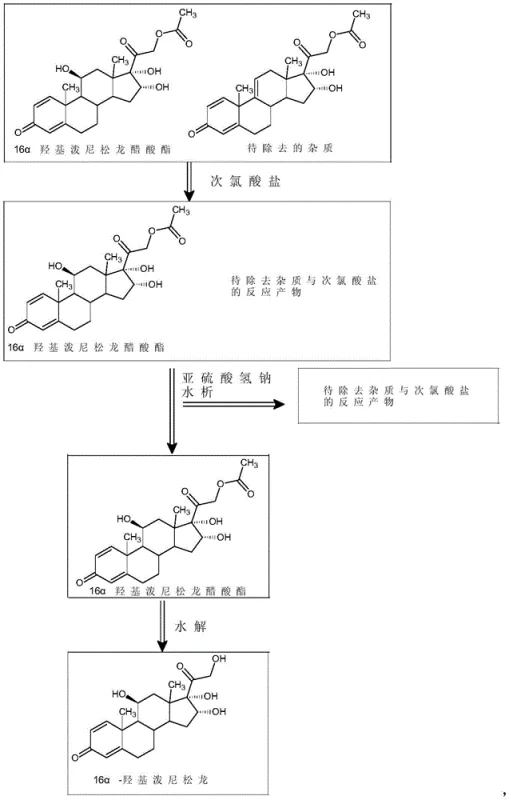 This solid-liquid separation is far more efficient than trying to separate two similar solids. Finally, the purified acetate undergoes hydrolysis using sodium sulfite in an alcohol solvent at low temperatures (-10°C to 10°C). This mild hydrolysis condition ensures the removal of the acetate group to reveal the free 21-hydroxyl without epimerizing the sensitive 16α-hydroxyl group, ultimately yielding the high-purity 16α-hydroxyprednisolone with a sharp melting point and excellent HPLC profile.
This solid-liquid separation is far more efficient than trying to separate two similar solids. Finally, the purified acetate undergoes hydrolysis using sodium sulfite in an alcohol solvent at low temperatures (-10°C to 10°C). This mild hydrolysis condition ensures the removal of the acetate group to reveal the free 21-hydroxyl without epimerizing the sensitive 16α-hydroxyl group, ultimately yielding the high-purity 16α-hydroxyprednisolone with a sharp melting point and excellent HPLC profile.
How to Synthesize 16α-Hydroxyprednisolone Efficiently
Implementing this synthesis route requires careful attention to solvent ratios and temperature control during the oxidation phase to maximize the selectivity of the impurity removal. The patent specifies using a mixed solvent system of dichloromethane and alcohols like methanol or ethanol, which provides the necessary solubility for the steroid while allowing effective interaction with the aqueous hypochlorite phase. Operators must strictly adhere to the dropwise addition of the oxidant to manage exotherms and maintain the reaction within the 15-40°C window.
- Dissolve crude 16α-hydroxyprednisolone acetate in a dichloromethane/alcohol mixture, add organic acid, and treat with aqueous hypochlorite at 15-40°C to modify impurities.
- Quench the reaction with sodium bisulfite, concentrate the solvent, and perform water precipitation to filter out the modified impurity byproducts.
- Hydrolyze the purified acetate intermediate in an organic solvent with sodium sulfite at low temperature (-10 to 10°C), followed by neutralization and crystallization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this purification technology offers substantial strategic benefits beyond mere technical superiority. The primary advantage lies in the drastic simplification of the purification train, which directly translates to reduced operational expenditures and improved throughput. By eliminating the need for multiple recrystallizations or preparative chromatography to remove Impurity H, manufacturers can significantly lower solvent consumption and waste disposal costs. Furthermore, the use of commodity chemicals like sodium hypochlorite and sodium bisulfite ensures that the supply chain for reagents remains stable and cost-effective, avoiding reliance on exotic or expensive catalysts that might introduce supply risks. The robustness of the process also means fewer batch failures due to off-spec purity, enhancing overall production reliability and ensuring consistent delivery schedules to downstream API clients.
- Cost Reduction in Manufacturing: The elimination of complex purification steps such as column chromatography or repeated recrystallizations leads to significant savings in both material and labor costs. By converting the impurity into a removable byproduct early in the sequence, the process maximizes the recovery of the valuable steroid intermediate, thereby improving the effective yield per kilogram of starting material. This efficiency gain allows for more competitive pricing structures for high-purity pharmaceutical intermediates without compromising on quality margins.
- Enhanced Supply Chain Reliability: The reliance on widely available, bulk-grade reagents like hypochlorites and common organic solvents mitigates the risk of raw material shortages that often plague specialty chemical supply chains. The simplicity of the operation also reduces the dependency on highly specialized equipment or skilled labor for complex separations, making it easier to scale production across different manufacturing sites. This flexibility ensures a continuous supply of critical intermediates even during periods of market volatility or logistical disruptions.
- Scalability and Environmental Compliance: The process operates under mild conditions and generates waste streams that are easier to treat compared to those containing heavy metal catalysts or hazardous organic byproducts. The ability to achieve high purity through chemical modification rather than physical separation reduces the volume of solvent waste generated per unit of product. This aligns well with modern green chemistry initiatives and environmental regulations, facilitating smoother regulatory approvals and reducing the environmental footprint of the manufacturing facility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this high-purity synthesis method. These insights are derived directly from the experimental data and process descriptions provided in the patent documentation, offering clarity on scalability and quality control measures.
Q: Why is Impurity H difficult to remove in conventional 16α-hydroxyprednisolone synthesis?
A: Impurity H possesses a chemical structure highly similar to the target product, leading to co-crystallization during standard recrystallization processes. Conventional purification methods often fail to separate them effectively, resulting in final products with impurity levels exceeding 0.90%, which fails stringent pharmaceutical specifications.
Q: How does the hypochlorite treatment improve product purity?
A: The novel method utilizes aqueous hypochlorite to selectively react with Impurity H under mild conditions (15-40°C). This chemical modification alters the polarity and solubility profile of the impurity, allowing it to remain in the mother liquor during subsequent water precipitation, thereby reducing impurity content to below 0.02%.
Q: What are the typical yield and purity metrics for this process?
A: Experimental data from the patent indicates that this optimized route achieves a total mass yield of approximately 71-73%. Crucially, the final product consistently demonstrates an HPLC purity greater than 99.5%, with Impurity H controlled to less than 0.02%, significantly outperforming traditional methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 16α-Hydroxyprednisolone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of purity and consistency in the production of corticosteroid intermediates like 16α-hydroxyprednisolone. Our technical team has extensively analyzed advanced purification technologies, including the hypochlorite-mediated route described in CN112125943A, to ensure our manufacturing capabilities meet the most stringent global standards. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, leveraging our state-of-the-art facilities and rigorous QC labs to guarantee batch-to-batch reproducibility. Our commitment to quality means we can deliver intermediates with purity specifications exceeding 99.5%, fully compliant with the demanding requirements of modern pharmaceutical formulations.
We invite potential partners to engage with our technical procurement team to discuss how our optimized manufacturing processes can support your supply chain needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our efficient synthesis routes can reduce your overall cost of goods. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project timelines, ensuring a seamless integration of our high-quality intermediates into your API production workflow.