Advanced Synthesis of Doxifluridine Intermediates Using Styryl-Benzoate Ribose Donors for Commercial Scale-Up
Introduction to Next-Generation Doxifluridine Synthesis
The global demand for effective antitumor agents continues to drive innovation in the synthesis of nucleoside analogues, with Doxifluridine standing out as a critical prodrug of 5-Fluorouracil used extensively in treating gastric and colorectal cancers. Recent intellectual property developments, specifically patent CN111072734A, have introduced a groundbreaking methodology for constructing the ribose-pyrimidine bond that forms the core of this molecule. This patent discloses a novel 5-deoxy-D-ribofuranose 1-[2-(1-styryl)benzoate] derivative which serves as a superior glycosyl donor compared to historical standards. By leveraging this specialized intermediate, manufacturers can achieve glycosylation under remarkably mild conditions, utilizing only catalytic amounts of activators rather than the harsh, stoichiometric reagents previously deemed necessary. This technological leap addresses long-standing challenges in purity and yield, positioning the industry for more efficient and cost-effective production of high-value oncology intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Doxifluridine has been plagued by significant chemical inefficiencies and operational hazards inherent to traditional glycosylation strategies. As illustrated in prior art schemes, conventional routes often rely on triacetyl deoxyribose reacting with silylated fluorouracil in the presence of equivalent or excessive amounts of strong Lewis acids such as stannic chloride or trimethylsilyl trifluoromethanesulfonate. These aggressive conditions frequently lead to severe side reactions, including the degradation of the sensitive fluorouracil base and the formation of difficult-to-remove anomeric mixtures. Furthermore, alternative pathways involving the functional group conversion of ribose-derived nucleosides are excessively lengthy, generating substantial chemical waste and suffering from poor overall atom economy. The incompatibility of these harsh acidic environments with various functional groups often necessitates complex protection and deprotection sequences, further inflating production costs and extending lead times for pharmaceutical manufacturers.
The Novel Approach
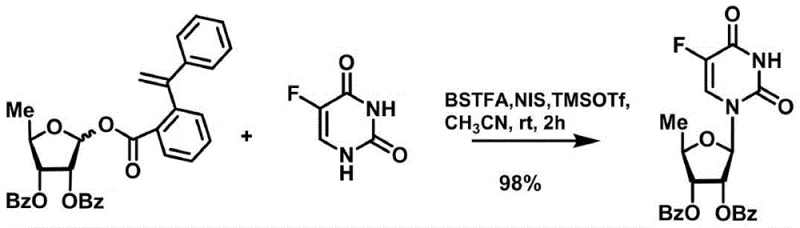
In stark contrast to these legacy methods, the innovative strategy outlined in CN111072734A utilizes a uniquely designed 5-deoxy-D-ribofuranose 1-[2-(1-styryl)benzoate] derivative to facilitate a highly selective coupling reaction. This novel glycosyl donor can be activated efficiently using a catalytic system comprising N-iodosuccinimide (NIS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf), completely eliminating the need for stoichiometric quantities of corrosive Lewis acids. The reaction proceeds smoothly at temperatures ranging from 0°C to room temperature, ensuring the structural integrity of the fluorouracil moiety is preserved throughout the synthesis. Experimental data from the patent indicates that this approach delivers an exceptional isolated yield of 98%, demonstrating near-quantitative conversion that drastically reduces the burden on downstream purification processes. This method represents a paradigm shift towards greener, more sustainable chemistry for the production of complex nucleoside pharmaceutical intermediates.
Mechanistic Insights into Catalytic Glycosylation Activation
The core mechanistic advantage of this synthesis lies in the specific activation of the anomeric center of the ribose donor through a synergistic catalytic cycle involving NIS and TMSOTf. In this system, the trimethylsilyl trifluoromethanesulfonate acts as a potent silylating agent that coordinates with the carbonyl oxygen of the benzoate leaving group, thereby increasing its electrophilicity and facilitating its departure. Simultaneously, N-iodosuccinimide serves as a source of electrophilic iodine, which likely interacts with the anomeric position to generate a highly reactive oxocarbenium ion intermediate or an alpha-iodo species in situ. This dual-activation mechanism ensures that the glycosyl donor is sufficiently reactive to couple with the silylated 5-fluorouracil nucleophile without requiring the extreme thermal or acidic conditions that typically degrade sensitive heterocyclic bases. The result is a highly stereoselective formation of the beta-N-glycosidic bond, which is crucial for the biological activity of the final Doxifluridine product.
From an impurity control perspective, the mildness of this catalytic system is instrumental in maintaining a clean reaction profile throughout the manufacturing process. Traditional methods employing excess Lewis acids often promote hydrolysis of the acetyl or benzoyl protecting groups prematurely, leading to a complex mixture of partially deprotected species that are notoriously difficult to separate via crystallization or chromatography. By contrast, the controlled generation of the reactive intermediate in this novel pathway minimizes such side reactions, ensuring that the protecting groups remain intact until the dedicated deprotection stage. This high level of chemoselectivity translates directly into a simplified isolation procedure, where the crude product requires minimal purification to meet stringent pharmaceutical specifications. Consequently, the overall process robustness is significantly enhanced, reducing the risk of batch failures and ensuring consistent quality for regulatory compliance.
How to Synthesize Doxifluridine Efficiently
The practical implementation of this advanced synthetic route involves a streamlined sequence of reactions designed for maximum operational simplicity and yield optimization. The process begins with the preparation of the key glycosyl donor, followed by the critical catalytic glycosylation step, and concludes with a straightforward deprotection to release the active pharmaceutical ingredient. Detailed procedural parameters, including specific solvent choices, temperature controls, and molar ratios, are essential for replicating the high success rates reported in the patent literature. Operators must ensure strict anhydrous conditions during the activation phase to prevent premature hydrolysis of the reactive intermediates. For a comprehensive understanding of the standardized operating procedures required to execute this synthesis effectively, please refer to the step-by-step guide provided below.
- Prepare the 5-deoxy-D-ribofuranose 1-[2-(1-styryl)benzoate] donor via esterification of the protected ribose with 2-(1-styryl)benzoic acid.
- Perform glycosylation by reacting the donor with silylated 5-fluorouracil using catalytic NIS and TMSOTf in acetonitrile.
- Execute final deprotection of the benzoyl groups using sodium methoxide in methanol to yield pure Doxifluridine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic methodology offers profound strategic benefits that extend far beyond simple chemical yield improvements. By transitioning from stoichiometric Lewis acid protocols to a catalytic activation system, manufacturing facilities can achieve significant reductions in raw material consumption and hazardous waste generation. The elimination of heavy metal catalysts like tin chloride removes the need for expensive and time-consuming metal scavenging steps, which are often a bottleneck in API production workflows. Furthermore, the mild reaction conditions reduce energy consumption associated with heating or cooling large-scale reactors, contributing to a lower overall carbon footprint for the manufacturing site. These factors collectively enhance the economic viability of producing Doxifluridine, making it a more attractive candidate for inclusion in generic drug portfolios and hospital formularies.
- Cost Reduction in Manufacturing: The shift to a catalytic process fundamentally alters the cost structure of nucleoside synthesis by removing the requirement for purchasing and disposing of large quantities of expensive Lewis acids. In traditional processes, the cost of reagents like TMSOTf or SnCl4, combined with the neutralization and disposal of their acidic by-products, constitutes a significant portion of the variable manufacturing costs. By utilizing these reagents in merely catalytic amounts, the new method drastically lowers the direct material costs per kilogram of produced intermediate. Additionally, the high 98% yield reported in the patent examples implies a substantial reduction in the loss of valuable starting materials, such as the fluorouracil base and the protected ribose donor. This efficiency gain means that less raw material is required to produce the same amount of final product, directly improving the gross margin for the manufacturer.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route contributes to a more resilient supply chain by minimizing the risks associated with batch variability and production delays. Conventional methods that rely on harsh conditions are prone to unexpected side reactions that can compromise entire batches, leading to shortages and delivery failures for downstream pharmaceutical customers. The mild and selective nature of the NIS/TMSOTf catalytic system ensures consistent reaction outcomes, allowing for more accurate production planning and inventory management. Moreover, the reagents involved in this new pathway, such as N-iodosuccinimide and standard organic solvents, are widely available from multiple global suppliers, reducing the risk of single-source dependency. This diversification of the supply base ensures continuity of supply even in the face of market fluctuations or geopolitical disruptions affecting specific chemical sectors.
- Scalability and Environmental Compliance: Scaling chemical processes from the laboratory to commercial production often exposes hidden safety and environmental hazards that are not apparent at small scales, but this methodology is inherently designed for safe scale-up. The avoidance of exothermic reactions driven by strong Lewis acids reduces the thermal load on reactors, lowering the risk of runaway reactions and enhancing operator safety in large-scale facilities. From an environmental compliance standpoint, the reduction in hazardous waste streams simplifies the permitting process for manufacturing sites and lowers the costs associated with wastewater treatment and solid waste disposal. The cleaner reaction profile also facilitates easier solvent recovery and recycling, aligning with modern green chemistry principles and corporate sustainability goals. These attributes make the technology highly attractive for contract development and manufacturing organizations (CDMOs) seeking to expand their capacity for oncology intermediates without incurring prohibitive infrastructure upgrade costs.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route for Doxifluridine. These answers are derived directly from the experimental data and technical disclosures found within the patent documentation to ensure accuracy and relevance for industry professionals. Understanding these details is crucial for R&D teams evaluating the feasibility of technology transfer and for procurement specialists assessing the long-term value proposition of this manufacturing method. We encourage stakeholders to review these insights carefully to inform their strategic decision-making processes regarding nucleoside sourcing and production.
Q: What is the primary advantage of the styryl-benzoate donor over traditional triacetyl deoxyribose?
A: The styryl-benzoate donor allows for activation under catalytic conditions using NIS and TMSOTf, avoiding the harsh, stoichiometric Lewis acids required by traditional methods, which significantly reduces side reactions and improves yield to 98%.
Q: How does this method improve impurity profiles for API manufacturing?
A: By utilizing mild reaction conditions (0°C to room temperature) and avoiding excessive Lewis acids, the process minimizes the degradation of the fluorouracil base and prevents the formation of complex by-products common in harsh acidic environments.
Q: Is this synthetic route suitable for large-scale commercial production?
A: Yes, the route is highly scalable due to the use of readily available reagents, simple workup procedures involving standard extraction and chromatography, and the elimination of hazardous stoichiometric metal catalysts.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Doxifluridine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic advancements described in CN111072734A and are fully equipped to leverage this technology for our global clientele. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless and efficient. Our state-of-the-art facilities are designed to handle complex nucleoside chemistry with the utmost precision, adhering to stringent purity specifications and rigorous QC labs that guarantee every batch meets international regulatory standards. We understand that consistency and quality are paramount in the pharmaceutical supply chain, and our commitment to excellence ensures that your project remains on track from initial development through to commercial launch.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis route can be tailored to your specific volume and quality requirements. By partnering with us, you gain access to a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this catalytic methodology for your specific supply chain context. We encourage you to contact us today to request specific COA data and route feasibility assessments that will demonstrate the tangible advantages of our manufacturing capabilities. Let us collaborate to optimize your supply of high-purity Doxifluridine intermediates and drive value across your entire oncology drug portfolio.