Advanced Catalytic Glycosylation for Commercial Scale-up of Doxifluridine Intermediates
Advanced Catalytic Glycosylation for Commercial Scale-up of Doxifluridine Intermediates
Introduction to Novel Doxifluridine Synthesis Technology
The pharmaceutical industry continuously seeks robust synthetic pathways for critical oncology agents, and the recent disclosure in patent CN111072734B presents a transformative approach to manufacturing Doxifluridine intermediates. This antitumor prodrug, which converts to 5-Fluorouracil within tumor tissues, requires precise stereochemical control during the glycosylation step to ensure efficacy and safety. Traditional methods often struggle with harsh reaction conditions that compromise yield and purity, creating significant bottlenecks for reliable API intermediate suppliers aiming for commercial viability. The innovation lies in the design of a specialized 5-deoxy-D-ribofuranose 1-[2-(1-styryl)benzoate] derivative that serves as a superior glycosyl donor. By leveraging this unique molecular architecture, manufacturers can achieve activation under remarkably mild conditions using catalytic amounts of trimethylsilyl trifluoromethanesulfonate and N-iodosuccinimide. This strategic shift away from stoichiometric reagents not only enhances the reaction efficiency but also aligns perfectly with the rigorous quality standards demanded by global regulatory bodies for high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
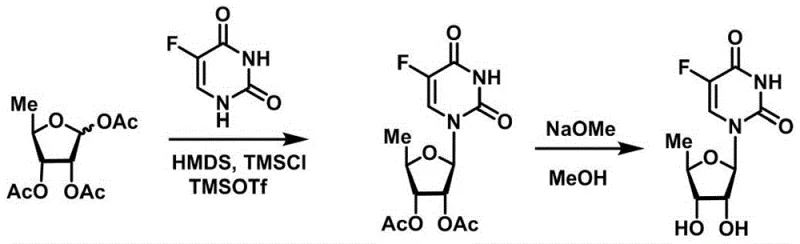
The Limitations of Conventional Methods
Historical synthetic routes for nucleoside analogues have predominantly relied on the use of equivalent or even excessive amounts of strong Lewis acids to drive the glycosylation reaction to completion. As illustrated in prior art schemes, methods utilizing reagents such as tin tetrachloride or stoichiometric trimethylsilyl trifluoromethanesulfonate often necessitate severe reaction conditions that are incompatible with many sensitive functional groups present in complex molecules. These harsh environments frequently lead to the formation of undesirable by-products and anomeric mixtures, complicating the downstream purification process and drastically reducing the overall material throughput. Furthermore, the reliance on heavy metal catalysts introduces significant challenges in meeting stringent residual metal specifications required for final drug substances, thereby increasing the cost and complexity of manufacturing. The inefficiency of these traditional protocols often results in prolonged reaction times and lower yields, which negatively impacts the economic feasibility for any cost reduction in pharmaceutical intermediates manufacturing initiative.
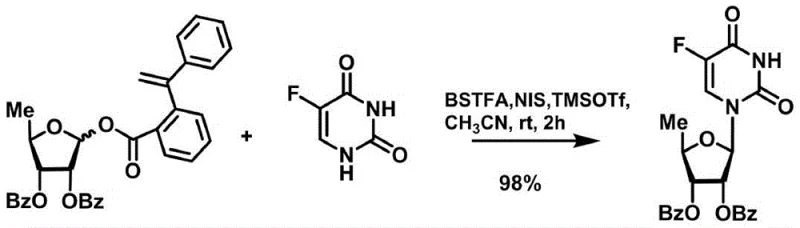
The Novel Approach
In stark contrast to legacy methodologies, the novel approach described in the patent utilizes a specifically engineered sugar donor that can be activated efficiently using only catalytic quantities of Lewis acid promoters. This breakthrough allows the glycosylation to proceed under mild temperatures, typically ranging from 0°C to room temperature, which preserves the integrity of the molecular structure and prevents degradation. The synergy between the styryl benzoate moiety and the catalytic system of N-iodosuccinimide and trimethylsilyl trifluoromethanesulfonate facilitates a highly selective formation of the desired beta-anomer with exceptional yields reaching up to 98%. Such high efficiency eliminates the need for extensive recycling of unreacted starting materials and significantly reduces the volume of chemical waste generated per kilogram of product. For procurement teams, this translates to a more predictable supply chain and reduced raw material consumption, while R&D directors benefit from a cleaner reaction profile that simplifies analytical characterization and quality control workflows.
Mechanistic Insights into Catalytic Glycosylation Activation
The core of this technological advancement resides in the unique electronic properties of the 1-[2-(1-styryl)benzoate] group attached to the anomeric position of the ribofuranose ring. Upon exposure to the catalytic system, the electron-rich styryl double bond interacts with the iodine species generated from N-iodosuccinimide, creating a highly reactive intermediate that facilitates the departure of the leaving group. This mechanism generates an oxocarbenium ion species that is sufficiently electrophilic to react with the silylated fluorouracil nucleophile but stable enough to prevent random decomposition or polymerization side reactions. The presence of the ortho-substituted benzoate further stabilizes the transition state through potential neighboring group participation or steric shielding, ensuring high stereoselectivity towards the therapeutically active anomer. Understanding this mechanistic pathway is crucial for process chemists aiming to optimize reaction parameters such as solvent polarity and addition rates to maximize the commercial scale-up of complex pharmaceutical intermediates without compromising on the critical quality attributes of the final active ingredient.
Impurity control is inherently built into this catalytic cycle due to the mildness of the activation conditions and the specificity of the reagent interactions. Unlike traditional strong acid methods that can cause acyl migration or sugar ring opening, this system maintains the structural fidelity of the furanose ring throughout the transformation. The use of catalytic rather than stoichiometric activators means that there are fewer acidic residues to neutralize and remove during the workup phase, leading to a simpler isolation procedure. Additionally, the high conversion rate minimizes the presence of unreacted sugar donor in the crude mixture, which reduces the burden on chromatographic purification steps. For supply chain heads, this reliability in impurity profiles ensures consistent batch-to-batch quality, reducing the risk of production delays caused by out-of-specification results and supporting the continuous delivery of high-purity pharmaceutical intermediates to downstream formulation partners.
How to Synthesize Doxifluridine Efficiently
The synthesis of Doxifluridine via this innovative route involves a streamlined sequence that begins with the preparation of the specialized sugar donor followed by the key glycosylation and final deprotection steps. The process is designed to be operationally simple, utilizing common organic solvents and reagents that are readily available in the global chemical market, thus avoiding supply bottlenecks associated with exotic catalysts. Detailed standard operating procedures for each stage, including precise molar ratios, temperature controls, and quenching protocols, are essential for reproducing the high yields reported in the patent literature. Manufacturers should pay close attention to the moisture content of the solvents and the activation of the nucleobase to ensure optimal performance of the catalytic system. The following guide outlines the critical phases of this synthesis, providing a roadmap for technical teams to implement this efficient methodology in their pilot or production facilities.
- Prepare the 5-deoxy-D-ribofuranose 1-[2-(1-styryl)benzoate] derivative through glycosylation, desulfurization, and condensation.
- Activate the sugar donor using catalytic TMSOTf and NIS in anhydrous acetonitrile with silylated fluorouracil.
- Perform alkaline hydrolysis using sodium methoxide in methanol to remove protecting groups and obtain Doxifluridine.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this novel synthetic route offers substantial strategic benefits for organizations focused on optimizing their manufacturing economics and supply chain resilience. The shift from stoichiometric to catalytic reagent usage fundamentally alters the cost structure of the process by reducing the consumption of expensive Lewis acids and simplifying the waste treatment requirements. This efficiency gain is particularly valuable in the context of rising raw material costs and increasingly strict environmental regulations governing chemical production. By minimizing the number of purification steps and improving the overall yield, the process enhances the throughput of existing manufacturing assets without requiring significant capital investment in new equipment. These factors collectively contribute to a more robust and cost-effective supply chain capable of meeting the growing global demand for oncology medications while maintaining competitive pricing structures.
- Cost Reduction in Manufacturing: The elimination of stoichiometric amounts of expensive Lewis acids and the reduction in solvent usage for purification directly lower the variable cost per kilogram of the produced intermediate. Furthermore, the high yield reduces the amount of starting material required to produce a fixed quantity of product, enhancing overall material efficiency. The simplified workup procedure also decreases labor hours and utility consumption associated with extended reaction times and complex separations. These cumulative effects result in significant cost savings that can be passed down the supply chain or reinvested into further process optimization initiatives.
- Enhanced Supply Chain Reliability: Utilizing readily available and stable reagents reduces the dependency on specialized suppliers who may face production disruptions or geopolitical constraints. The mild reaction conditions lower the safety risks associated with handling hazardous chemicals, ensuring smoother operations and fewer unplanned shutdowns due to safety incidents. Consistent high yields and predictable reaction outcomes allow for more accurate production planning and inventory management, reducing the need for large safety stocks. This reliability strengthens the partnership between the manufacturer and their clients, ensuring a steady flow of critical materials for drug development and commercial launch.
- Scalability and Environmental Compliance: The process is inherently scalable due to its exothermic profile being manageable under standard cooling capacities and the absence of highly toxic heavy metal residues. Reduced chemical waste generation aligns with green chemistry principles, lowering the environmental footprint and simplifying compliance with local and international environmental protection laws. The ease of scaling from laboratory to commercial production minimizes the technical risks typically associated with technology transfer, accelerating the time to market for new generic or branded formulations. This sustainability aspect also enhances the corporate reputation of the manufacturer among stakeholders who prioritize environmental responsibility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this advanced synthesis technology for Doxifluridine production. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the practical advantages of this method. Understanding these details helps decision-makers evaluate the feasibility of adopting this route for their specific manufacturing needs and regulatory contexts. The information covers aspects ranging from reaction mechanics to supply chain implications, offering a comprehensive overview for technical and commercial stakeholders alike.
Q: What is the primary advantage of the new glycosylation method over traditional routes?
A: The new method utilizes catalytic amounts of Lewis acid instead of stoichiometric equivalents, resulting in milder reaction conditions and significantly higher yields up to 98%.
Q: How does this process improve impurity control for pharmaceutical grades?
A: By avoiding harsh acidic conditions and excess metal catalysts, the process minimizes side reactions and simplifies the removal of residual metals, ensuring high purity specifications.
Q: Is this synthetic route suitable for large-scale manufacturing?
A: Yes, the use of readily available reagents, mild temperatures, and simplified workup procedures makes this route highly scalable for commercial production of API intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Doxifluridine Supplier
At NINGBO INNO PHARMCHEM, we leverage deep technical expertise to transform innovative patent technologies into commercially viable manufacturing processes for our global clientele. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of high-yield catalytic glycosylation are fully realized in large-scale operations. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Doxifluridine intermediate meets the highest international pharmacopoeia standards. Our commitment to quality and consistency makes us a trusted partner for pharmaceutical companies seeking to secure their supply chains for critical oncology ingredients.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis route can be tailored to your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain detailed insights into the potential economic benefits of switching to this catalytic method for your production needs. We encourage you to contact us to obtain specific COA data and route feasibility assessments that demonstrate our capability to deliver high-quality intermediates reliably. Let us collaborate to optimize your supply chain and accelerate the delivery of life-saving medications to patients worldwide.