Advanced Halogen-Free Synthesis of C-Nucleoside Derivatives for Commercial Antiviral Production
Introduction to Breakthrough C-Nucleoside Synthesis Technology
The global demand for effective antiviral therapeutics has placed immense pressure on the pharmaceutical supply chain to deliver high-purity active pharmaceutical ingredients (APIs) with greater efficiency and reliability. In this context, Chinese Patent CN113248508A represents a significant technological leap forward in the synthesis of C-nucleoside derivatives, which serve as critical precursors for drugs such as Remdesivir. This patent discloses a novel methodology that fundamentally alters the traditional synthetic landscape by utilizing N-protected heterocyclic compounds to prepare C-nucleoside derivatives without the need for halogenation or temporary amino protection. By streamlining the synthetic route and enhancing reaction yields, this technology addresses key bottlenecks in the manufacturing of complex nucleoside analogs. For R&D directors and procurement strategists, understanding the implications of this halogen-free approach is essential for optimizing supply chains and reducing the cost of goods sold (COGS) in antiviral drug manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of C-nucleoside analogs has relied heavily on methodologies reported by major pharmaceutical entities, which often involve multi-step protection and deprotection sequences that introduce significant inefficiencies. For instance, earlier methods described by Gilead Sciences required the temporary silicon protection of heterocyclic bases followed by halogenation (bromination or iodination) to facilitate coupling with ribolactones. These conventional routes are fraught with challenges, including the use of expensive halogenated raw materials, the generation of hazardous waste, and the inherent instability of halogenated intermediates which can lead to variable reaction outcomes. Furthermore, the necessity for temporary amino protection adds additional synthetic steps, increasing the overall processing time and reducing the cumulative yield of the final product. In some reported instances, the yield from the halogenated base to the C-nucleoside analog was less than 40%, creating substantial material loss and driving up production costs for commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
The innovative strategy outlined in Patent CN113248508A circumvents these historical limitations by employing N-benzyloxycarbonyl (Cbz) or N-tert-butoxycarbonyl (Boc) protected heterocyclic compounds as the starting point for coupling reactions. This approach eliminates the need for halogenation entirely, allowing for the direct removal of protons from the heterocyclic ring using organolithium or organomagnesium reagents. The resulting nucleophilic species then undergoes addition with ribolactone to form the C-glycosidic bond directly. This streamlined pathway not only shortens the synthetic route by removing the halogenation and temporary protection steps but also significantly improves the reaction yield between the heterocyclic compound and the ribolactone. By avoiding the introduction of halogen atoms as substituents, the process enhances the stability of intermediates and simplifies the purification workflow, offering a robust alternative for reliable pharmaceutical intermediate supplier networks seeking to optimize their manufacturing protocols.
Mechanistic Insights into Organometallic Coupling and N-Protection
The core mechanistic advantage of this technology lies in the strategic use of N-protecting groups to modulate the reactivity and physicochemical properties of the heterocyclic base during the coupling phase. In the novel process, the amino group of the pyrrolo[2,1-f][1,2,4]triazine is first protected with a Boc or Cbz group, which serves a dual purpose: it prevents unwanted side reactions at the nitrogen center and modifies the solubility profile of the molecule. When treated with strong bases such as n-butyllithium or Turbo Grignard reagents (e.g., i-PrMgCl·LiCl), the N-protected heterocycle undergoes regioselective deprotonation at the carbon position adjacent to the nitrogen, generating a highly reactive organometallic intermediate. This species then attacks the carbonyl carbon of the ribolactone (Formula A), forming a hemiketal intermediate that is subsequently converted into the C-nucleoside structure. The presence of the lipophilic protecting group ensures that the intermediate remains soluble in organic solvents like tetrahydrofuran (THF) or dichloromethane (DCM), facilitating homogeneous reaction conditions that are critical for high conversion rates.
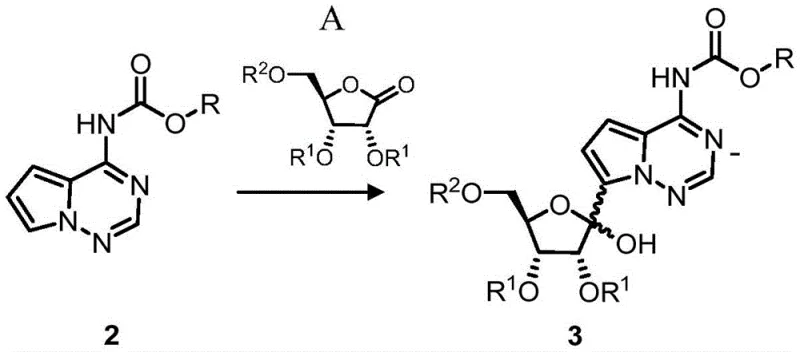
Furthermore, the retention of the N-protecting group through the subsequent cyanation and partial deprotection steps provides a distinct advantage in impurity control. Unlike unprotected amines which can exhibit high polarity and strong interactions with silica gel or other stationary phases, the Boc or Cbz protected intermediates display reduced polarity. This characteristic allows for more efficient separation of the desired product from reaction byproducts and unreacted starting materials during chromatographic purification. The ability to maintain the protecting group until the later stages of synthesis ensures that the stereochemical integrity of the C-glycosidic bond is preserved while minimizing the formation of N-glycosidic byproducts. This mechanistic refinement results in a cleaner crude product profile, reducing the burden on downstream purification processes and ensuring that the final API meets stringent purity specifications required for regulatory approval.
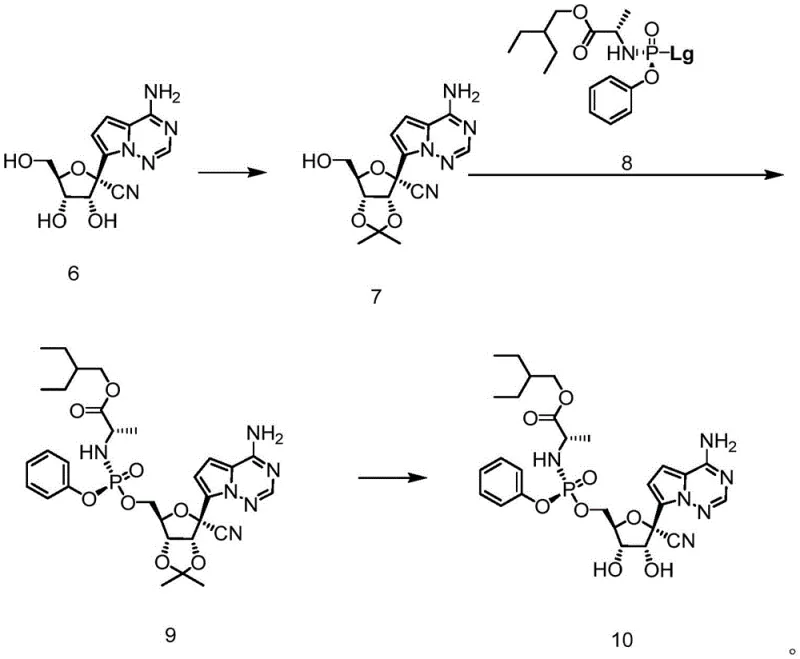
How to Synthesize C-Nucleoside Derivatives Efficiently
The implementation of this synthesis route requires precise control over reaction conditions, particularly regarding temperature and reagent stoichiometry, to maximize the efficiency of the organometallic coupling and subsequent functional group transformations. The process begins with the protection of the heterocyclic amine, followed by the critical lithiation or magnesiation step which must be performed at low temperatures (typically between -78°C and -50°C) to prevent decomposition of the organometallic species. Following the coupling with ribolactone, the hydroxyl group is substituted with a cyano group using reagents such as TMSCN or ZnCN in the presence of Lewis acids like TMSOTf. The detailed standardized synthesis steps for executing this protocol are provided in the guide below.
- Perform amino protection on the heterocyclic compound (Formula 1) using Boc2O or CbzCl to obtain the N-protected intermediate (Formula 2).
- React the N-protected heterocycle with ribolactone (Formula A) using organolithium or organomagnesium reagents to form the coupled product (Formula 3).
- Execute substitution of the hydroxyl group with a cyano group followed by deprotection to yield the final C-nucleoside derivative (Formula 6).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this halogen-free synthesis method offers tangible benefits that extend beyond mere technical feasibility, directly impacting the bottom line and operational resilience of antiviral drug production. By eliminating the requirement for halogenated starting materials, which are often subject to price volatility and supply constraints, manufacturers can achieve significant cost reduction in pharmaceutical intermediates manufacturing. The removal of these expensive reagents not only lowers the direct material costs but also reduces the complexity of waste disposal, as halogenated organic waste requires specialized treatment protocols. Additionally, the shortened synthetic route translates to reduced processing time and lower utility consumption per kilogram of product, contributing to a more sustainable and economically viable production model.
- Cost Reduction in Manufacturing: The elimination of halogenation steps and temporary silicon protection groups removes the need for costly reagents such as brominating agents and silyl chlorides, which are significant cost drivers in traditional nucleoside synthesis. Furthermore, the improved yield obtained through this direct coupling method means that less raw material is required to produce the same amount of final product, effectively lowering the cost per unit. The simplified purification process, driven by the favorable solubility of N-protected intermediates, also reduces the consumption of chromatography media and solvents, leading to substantial cost savings in downstream processing operations.
- Enhanced Supply Chain Reliability: Relying on non-halogenated starting materials diversifies the supplier base and mitigates the risk of shortages associated with specialized halogenated chemicals. The robustness of the organolithium and organomagnesium coupling reactions ensures consistent batch-to-batch quality, which is crucial for maintaining continuous production schedules. By adopting a synthesis route that is less sensitive to the variability of halogenated precursors, supply chain leaders can secure a more stable flow of critical intermediates, thereby reducing lead time for high-purity pharmaceutical intermediates and ensuring uninterrupted availability for API formulation.
- Scalability and Environmental Compliance: The streamlined nature of this process, characterized by fewer unit operations and the absence of hazardous halogenation steps, makes it inherently more scalable for industrial production. The reduction in hazardous waste generation aligns with increasingly strict environmental regulations, minimizing the regulatory burden and potential liabilities associated with chemical manufacturing. This environmental compliance, combined with the operational simplicity of the route, facilitates easier technology transfer from laboratory to pilot and commercial scales, ensuring that the supply chain can rapidly respond to surges in demand for antiviral therapies without compromising on safety or quality standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel C-nucleoside synthesis technology, providing clarity on its practical application and benefits for industry stakeholders. These insights are derived directly from the technical disclosures within the patent documentation to ensure accuracy and relevance for decision-makers evaluating this process for integration into their manufacturing portfolios.
Q: How does this new synthesis method improve upon conventional Gilead routes?
A: Unlike conventional routes that require temporary silicon protection and halogenation of the heterocyclic base, this method utilizes direct N-protection (Boc/Cbz). This eliminates the need for expensive halogenated starting materials and reduces the total number of synthetic steps, leading to higher overall yields.
Q: What are the purification advantages of using N-protected intermediates?
A: The presence of lipophilic protecting groups like tert-butyloxycarbonyl (Boc) or benzyloxycarbonyl (Cbz) significantly reduces the polarity of intermediate compounds. This enhanced solubility in organic solvents facilitates easier crystallization and chromatographic purification, resulting in higher purity final products.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process is designed for scalability. By removing complex halogenation steps and utilizing robust organometallic coupling conditions, the method offers improved operational simplicity and repeatability, which are critical factors for successful commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Remdesivir Intermediate Supplier
The technological advancements detailed in Patent CN113248508A underscore the potential for more efficient and cost-effective production of critical antiviral intermediates, yet realizing this potential requires a manufacturing partner with deep technical expertise and proven scale-up capabilities. NINGBO INNO PHARMCHEM stands at the forefront of this evolution, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, ensuring that every batch of C-nucleoside intermediate delivered meets the highest standards of quality and consistency required by global regulatory bodies. We understand the complexities of organometallic chemistry and the precise control needed for successful implementation of these advanced synthetic routes.
We invite pharmaceutical companies and contract manufacturing organizations to collaborate with us to leverage this innovative synthesis technology for their antiviral drug pipelines. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific production volumes and quality requirements. We encourage you to reach out today to obtain specific COA data and route feasibility assessments that demonstrate how our optimized manufacturing processes can enhance your supply chain efficiency and reduce overall production costs while ensuring a reliable supply of high-quality pharmaceutical intermediates.