Advanced Manufacturing of Tenofovir Alafenamide Intermediates via Optimized Phosphorylation
The global demand for effective antiretroviral therapies continues to drive innovation in the synthesis of nucleotide analogs, specifically within the realm of HIV treatment protocols. Patent CN109942633B, published in late August 2021, discloses a highly efficient preparation method for a critical intermediate of Tenofovir Alafenamide (TAF), known chemically as (R)-9-[2-(phosphoryl phenol methoxyl) propyl] adenine. This intermediate serves as the foundational building block for TAF hemifumarate, a prodrug that has demonstrated superior antiviral capability and safety profiles compared to earlier generations of tenofovir disoproxil fumarate (TDF). The structural complexity of the final active pharmaceutical ingredient necessitates a robust and scalable synthetic route for its precursor, ensuring that supply chains can meet the rigorous quality standards required for clinical and commercial applications.
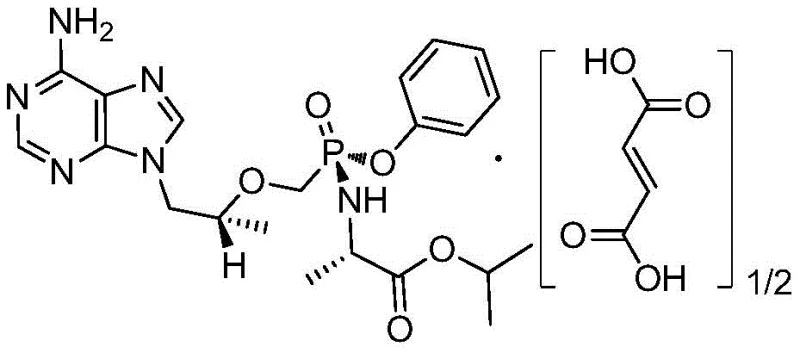
As a leading entity in the fine chemical sector, understanding the nuances of this patented methodology is essential for stakeholders aiming to secure a reliable pharmaceutical intermediate supplier. The disclosed technology addresses long-standing inefficiencies in the manufacturing landscape, offering a pathway that balances high yield with operational simplicity. By optimizing reaction conditions and solvent systems, this method not only enhances the economic viability of production but also aligns with modern green chemistry principles by minimizing waste generation. For R&D directors and procurement specialists alike, the adoption of such advanced synthetic strategies represents a strategic opportunity to optimize cost structures while maintaining the highest standards of product integrity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations detailed in CN109942633B, the synthesis of this key adenine derivative was plagued by several significant technical and economic bottlenecks that hindered large-scale industrial adoption. For instance, earlier methodologies described in documents like WO2013052094 relied heavily on acetonitrile as a solvent, requiring excessive volumes up to 8 times the mass of the reactants, which drastically inflated raw material costs and energy consumption for solvent recovery. Furthermore, these legacy processes often necessitated prolonged reaction times exceeding 48 hours under reflux conditions, creating substantial throughput limitations for manufacturing facilities. Alternative routes utilizing condensing agents such as dicyclohexylcarbodiimide (DCC) introduced severe downstream processing challenges, generating large quantities of difficult-to-remove urea byproducts and inorganic salts that compromised final product purity.
Additionally, methods employing thionyl chloride to activate phosphate groups, as seen in WO2002008041, imposed stringent equipment requirements due to the need for strictly anhydrous conditions and the handling of hazardous corrosive reagents. These conventional approaches collectively resulted in low atom economy, complex post-treatment procedures involving multiple extraction and filtration steps, and inconsistent yields that failed to meet the rigorous demands of modern API manufacturing. The reliance on nuclear magnetic resonance for process monitoring in some older protocols further added to the operational complexity and cost, making these routes unsuitable for the agile, cost-sensitive environment of contemporary pharmaceutical production.
The Novel Approach
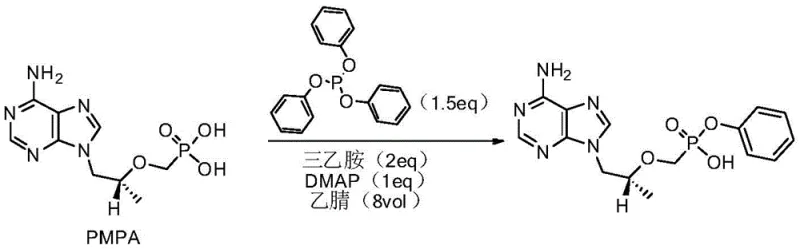
In stark contrast to these cumbersome legacy techniques, the novel approach presented in the patent utilizes a streamlined condensation reaction between (R)-9-[2-(phosphoryl methoxy) propyl] adenine (PMPA) and triphenyl phosphite within an N,N-Dimethylacetamide (DMA) solvent system. This strategic shift in solvent choice allows for a dramatic reduction in solvent volume, with optimal ratios ranging from merely 1 mL/g to 5 mL/g, thereby significantly concentrating the reaction mixture and improving thermal efficiency. The process operates effectively across a flexible temperature window, allowing manufacturers to choose between moderate temperatures for longer durations or higher temperatures for rapid conversion, providing unparalleled operational flexibility. By eliminating the need for hazardous activating agents like thionyl chloride and avoiding the formation of solid urea byproducts, this method simplifies the workflow to a single pot reaction followed by a straightforward precipitation step.
Mechanistic Insights into Triphenyl Phosphite-Mediated Condensation
The core of this technological breakthrough lies in the optimized interaction between the phosphonic acid moiety of PMPA and the triphenyl phosphite reagent, facilitated by a catalytic amount of 4-dimethylamino pyridine (DMAP) and an organic base such as triethylamine or DIPEA. In this mechanism, the base serves to deprotonate the phosphonic acid, enhancing its nucleophilicity, while the DMAP catalyst likely activates the phosphite species towards nucleophilic attack, promoting the formation of the phosphonate ester bond with high selectivity. The use of DMA as a polar aprotic solvent is critical, as it effectively solubilizes both the polar starting materials and the transition states involved, stabilizing the reaction intermediates and preventing premature precipitation or side reactions that could lead to impurity formation. This careful balancing of reactivity ensures that the condensation proceeds cleanly to form the desired phenol ester linkage without degradation of the sensitive adenine ring system.
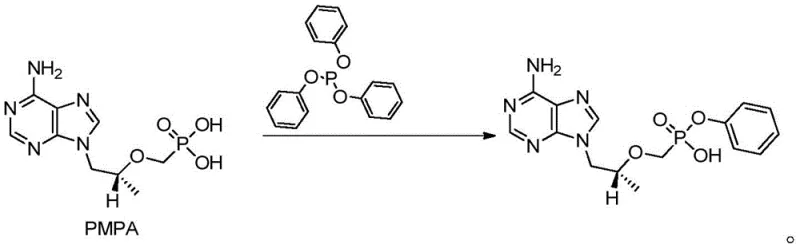
Impurity control is inherently built into this mechanistic design through the precise regulation of reaction parameters and the simplicity of the workup procedure. By monitoring the reaction progress via HPLC until the starting PMPA content drops below 0.1%, manufacturers can ensure near-quantitative conversion, minimizing the presence of unreacted starting materials in the crude mixture. The subsequent workup involves a pH-adjusted precipitation where the product selectively crystallizes out of the aqueous phase upon acidification to pH 2-4, leaving soluble organic impurities and catalyst residues in the mother liquor. This physical separation mechanism is far superior to chromatographic purifications or complex salt formations, resulting in a final product with HPLC purity consistently exceeding 99%, which is essential for meeting the stringent specifications required for downstream API synthesis.
How to Synthesize (R)-9-[2-(phosphoryl phenol methoxyl) propyl] adenine Efficiently
Implementing this synthesis requires strict adherence to the optimized molar ratios and thermal profiles established in the patent examples to ensure reproducibility and safety. The process begins with the charging of PMPA, the catalyst, the phosphite reagent, and the base into the DMA solvent, followed by a controlled heating phase that drives the condensation to completion. Operators must utilize standard analytical techniques such as HPLC to determine the exact endpoint, ensuring that the reaction is neither quenched prematurely nor allowed to run unnecessarily long, which could degrade product quality. The following guide outlines the standardized operational framework derived directly from the patent's preferred embodiments, serving as a blueprint for scaling this technology from laboratory benchtop to commercial production vessels.
- Charge (R)-9-[2-(phosphoryl methoxy) propyl] adenine (PMPA), 4-dimethylamino pyridine (DMAP), triphenyl phosphite, and an organic base (Triethylamine or DIPEA) into N,N-Dimethylacetamide (DMA) solvent.
- Heat the reaction mixture to a temperature between 95°C and 140°C and maintain stirring for a duration ranging from 3 to 26 hours, depending on the specific temperature profile selected.
- Upon completion, cool the mixture, extract with dichloromethane, and adjust the aqueous phase pH to 2-4 using concentrated hydrochloric acid to precipitate the pure product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented methodology translates into tangible strategic advantages that extend far beyond simple yield improvements. The drastic reduction in solvent volume directly correlates to a significant decrease in raw material procurement costs and a smaller environmental footprint, as less solvent needs to be purchased, stored, and ultimately distilled or disposed of. Furthermore, the elimination of expensive and hazardous reagents like thionyl chloride and DCC removes the need for specialized corrosion-resistant equipment and complex waste treatment protocols, thereby lowering capital expenditure and operational overheads. The simplified post-treatment process, which relies on precipitation rather than column chromatography or extensive recrystallization, drastically reduces labor hours and processing time, allowing for faster batch turnover and improved facility utilization rates.
- Cost Reduction in Manufacturing: The shift to a low-volume DMA solvent system and the use of catalytic amounts of DMAP significantly lowers the bill of materials compared to traditional methods that require stoichiometric amounts of coupling agents. By avoiding the generation of solid byproducts like dicyclohexylurea, the process eliminates costly filtration and washing steps, leading to substantial savings in utility consumption and waste disposal fees. The overall atom economy is markedly improved, ensuring that a higher percentage of input mass is converted into valuable product rather than waste, which is a critical factor in maintaining competitive pricing in the generic pharmaceutical market.
- Enhanced Supply Chain Reliability: All reagents utilized in this process, including triphenyl phosphite, PMPA, and common organic bases, are commercially available commodities with stable supply chains, reducing the risk of production delays due to raw material shortages. The robustness of the reaction conditions, which tolerate a range of temperatures and times without significant loss of yield, provides a buffer against minor operational variances, ensuring consistent output quality even in large-scale manufacturing environments. This reliability allows supply chain planners to forecast production timelines with greater accuracy, minimizing the need for safety stock and enabling a more lean and responsive inventory management strategy.
- Scalability and Environmental Compliance: The absence of hazardous gas evolution and the use of standard organic solvents make this process highly scalable from pilot plant to multi-ton production without requiring exotic reactor configurations. The simplified workup generates less aqueous and organic waste, facilitating easier compliance with increasingly stringent environmental regulations and reducing the burden on effluent treatment plants. This alignment with green chemistry principles not only mitigates regulatory risk but also enhances the corporate sustainability profile, which is becoming an increasingly important criterion for partnerships with major multinational pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route, derived from a detailed analysis of the patent's experimental data and comparative examples. Understanding these nuances is vital for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the long-term viability of this supply source. The answers provided reflect the specific advantages and operational parameters disclosed in the intellectual property, ensuring that expectations are aligned with the proven capabilities of the method.
Q: Why is N,N-Dimethylacetamide (DMA) preferred over Acetonitrile for this synthesis?
A: According to patent CN109942633B, using DMA significantly reduces solvent consumption compared to traditional acetonitrile methods which require up to 8 volumes. DMA allows for a more concentrated reaction environment (1-5 mL/g), leading to simplified post-treatment and reduced energy costs for solvent recovery.
Q: How does this method improve reaction time compared to prior art?
A: Traditional methods often require refluxing for over 48 hours. This novel approach offers flexibility: at lower temperatures (95-110°C), the reaction completes in roughly 20-26 hours, while at elevated temperatures (130-140°C), the reaction time is drastically shortened to just 2-4 hours without compromising yield.
Q: What represents the primary advantage regarding product purity?
A: The process utilizes a simple acid-base workup involving pH adjustment to 2-4, which effectively precipitates the product while leaving impurities in solution. This avoids the complex purification steps associated with DCC-mediated couplings, consistently achieving HPLC purity levels exceeding 99%.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tenofovir Alafenamide Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of life-saving antiretrovirals depends on the availability of high-quality intermediates produced via robust and scalable pathways. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the volumetric demands of global API manufacturers without compromising on quality. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify that every batch of (R)-9-[2-(phosphoryl phenol methoxyl) propyl] adenine meets or exceeds the purity profiles outlined in patent CN109942633B. Our commitment to technical excellence ensures that our clients receive a product that seamlessly integrates into their downstream synthesis processes, minimizing deviations and maximizing final API yield.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can be tailored to your specific supply chain requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this novel manufacturing protocol. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that will enhance the efficiency and profitability of your pharmaceutical manufacturing operations.