Advanced Semaglutide Manufacturing: Native Chemical Ligation and Desulfurization Strategy
Advanced Semaglutide Manufacturing: Native Chemical Ligation and Desulfurization Strategy
The pharmaceutical industry's demand for GLP-1 analogs like Semaglutide has necessitated the development of robust, scalable synthetic routes that ensure high purity and cost-efficiency. Patent CN114685645A introduces a transformative methodology for Semaglutide production, leveraging Native Chemical Ligation (NCL) combined with a selective desulfurization protocol. This approach addresses the inherent challenges of synthesizing long-chain peptides, specifically the issues of resin shrinkage and deletion sequences associated with traditional stepwise Solid-Phase Peptide Synthesis (SPPS). By dividing the synthesis into manageable fragments—specifically utilizing a Cysteine surrogate at position 18 or 19—the process enables the convergent assembly of the full peptide chain in the liquid phase. This strategic fragmentation not only mitigates the risk of racemization but also significantly simplifies the purification landscape, positioning this technology as a vital asset for reliable API intermediate suppliers aiming to optimize their manufacturing pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis of complex peptides like Semaglutide often relies on linear Fmoc-SPPS, which faces severe limitations as the chain length increases. A primary technical bottleneck is the physical shrinkage of the resin matrix during repeated coupling and deprotection cycles, which drastically reduces solvent accessibility to the reactive sites. This phenomenon leads to incomplete couplings and the formation of deletion peptides—impurities that lack one or more amino acids and possess physicochemical properties nearly identical to the target molecule. Consequently, the final crude product contains a complex mixture of closely related impurities, making downstream purification via preparative HPLC extremely difficult, costly, and yield-limiting. Furthermore, the direct synthesis of peptide thioesters, essential for NCL, is problematic because the thioester bond is labile to the piperidine conditions required for Fmoc group removal, rendering standard protocols ineffective for generating the necessary N-terminal thioester fragments.
The Novel Approach
The methodology disclosed in the patent circumvents these obstacles through a sophisticated fragment condensation strategy that decouples the synthesis of the N-terminal and C-terminal domains. Instead of attempting a linear build-up, the process synthesizes two distinct peptide fragments separately: an N-terminal thioester fragment (Fragment I or II) and a C-terminal fragment containing a Cysteine residue (Fragment III or IV). The innovation lies in the use of a "side-chain anchoring" technique for the thioester fragment, where the peptide is attached to the resin via the side chain of a Glutamic acid residue protected with an Allyl group. This allows the main chain carboxyl group to remain available for conversion into a thioester after the selective removal of the Allyl protecting group, thereby preserving the integrity of the thioester functionality throughout the SPPS process. This convergent approach ensures that each fragment can be purified individually before ligation, drastically reducing the complexity of the final crude mixture.
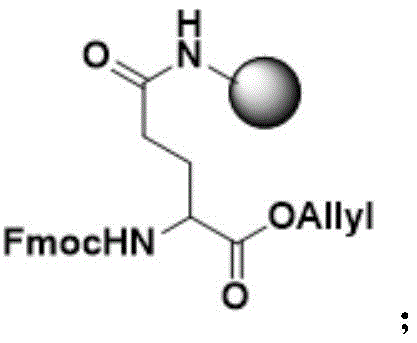
Mechanistic Insights into Side-Chain Anchored Thioester Synthesis
The core chemical innovation enabling this route is the strategic use of orthogonal protecting groups to facilitate thioester formation on solid support. As illustrated in the synthesis of Fragment I, the peptide chain is assembled on a resin where the anchoring point is the side-chain carboxyl of a Glutamic acid residue, initially protected as an Allyl ester (Fmoc-Glu-OAllyl). This orthogonal protection scheme is critical because the Allyl group is stable to the piperidine used for Fmoc deprotection but can be selectively removed under mild palladium-catalyzed conditions (using Pd(PPh3)4 and Morpholine) without affecting the rest of the peptide chain or the resin linkage. Once the Allyl group is cleaved, the free alpha-carboxyl group is exposed and immediately activated for nucleophilic attack by a thiol (such as ethyl 3-mercaptopropionate), forming the desired peptide thioester directly on the solid phase. This mechanism effectively bypasses the instability of thioesters to basic conditions, allowing for the robust generation of high-quality thioester fragments ready for ligation.
Following the ligation of the two fragments, the process employs a radical-induced desulfurization to convert the temporary Cysteine residue back into the native Alanine found in the Semaglutide sequence. This step is performed in a one-pot manner immediately after the NCL reaction reaches completion. The reaction system utilizes a radical initiator (VA-044) in the presence of a reducing agent (TCEP·HCl) and a hydrogen donor (such as reduced glutathione or MESNa). The mechanism involves the generation of thiyl radicals from the Cysteine thiol, which abstract a hydrogen atom from the beta-carbon, ultimately replacing the sulfur atom with hydrogen. This transformation is highly chemoselective and proceeds efficiently in the same buffer system used for ligation, eliminating the need for intermediate isolation and purification. The result is a seamless transition from the ligated intermediate to the final Semaglutide backbone with minimal epimerization or side reactions.
How to Synthesize Semaglutide Efficiently
The implementation of this synthetic route requires precise control over reaction conditions to maximize yield and purity, particularly during the liquid-phase ligation step. The process begins with the independent preparation of the purified peptide fragments using the solid-phase strategies described above. Once the fragments are obtained, they are dissolved in a specialized ligation buffer containing high concentrations of Guanidine Hydrochloride and, crucially, Imidazole. The reaction is allowed to proceed at room temperature, facilitating the chemoselective coupling between the thioester and the cysteine thiol. Upon confirmation of ligation completion via HPLC, the desulfurization reagents are added directly to the reaction vessel. This telescoped operation minimizes material handling and loss, streamlining the workflow from fragmented precursors to the full-length crude peptide. For detailed standardized operating procedures and specific reagent quantities, please refer to the technical guide below.
- Synthesize peptide thioester fragments (Fragment I or II) using a side-chain anchored Glu-OAllyl strategy on solid phase to prevent racemization.
- Prepare hydrophobic C-terminal fragments (Fragment III or IV) containing a Cysteine surrogate at the ligation site using Wang resin.
- Perform one-pot native chemical ligation in imidazole buffer followed by radical-induced desulfurization to convert Cysteine to Alanine.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic methodology offers substantial advantages in terms of cost structure and supply chain reliability for large-scale API manufacturing. The ability to synthesize fragments in parallel significantly compresses the overall production timeline compared to linear synthesis, allowing for a more responsive supply chain capable of meeting fluctuating market demands. Moreover, the purification profile of the crude product is markedly improved; the primary impurities are uncondensed starting fragments rather than deletion peptides. These unreacted fragments are chemically distinct from the final product, making them easier to separate via chromatography and, importantly, allowing for their recovery and recycling. This recyclability translates directly into raw material cost savings, as expensive amino acid derivatives are not lost to waste streams but can be reintroduced into the process.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts in the final desulfurization step (using organic radical initiators instead) removes the need for expensive and time-consuming heavy metal scavenging processes, which are often required to meet strict regulatory limits for parenteral drugs. Furthermore, the high efficiency of the ligation reaction reduces the excess of reagents needed to drive the reaction to completion, lowering the consumption of high-cost peptide fragments. The simplified purification workflow also reduces the burden on preparative HPLC columns and solvents, leading to significant operational expenditure reductions in the downstream processing phase.
- Enhanced Supply Chain Reliability: By breaking the synthesis into smaller, modular fragments, the manufacturing process becomes more resilient to bottlenecks. If a specific amino acid coupling encounters issues, it affects only a small fragment rather than the entire 31-residue chain, reducing the risk of total batch failure. This modularity facilitates better inventory management of intermediates, allowing manufacturers to stockpile purified fragments and rapidly assemble the final API as needed. This flexibility is crucial for maintaining continuity of supply in the face of raw material volatility or unexpected surges in demand for GLP-1 therapies.
- Scalability and Environmental Compliance: The use of Imidazole as a solubility enhancer and catalyst avoids the need for harsh organic co-solvents that might be required to dissolve highly hydrophobic fragments, aligning the process with greener chemistry principles. The one-pot nature of the ligation-desulfurization sequence minimizes solvent usage and waste generation associated with intermediate workups. Additionally, the robustness of the solid-phase thioester synthesis ensures consistent quality across different batch sizes, facilitating a smoother scale-up from pilot plant to commercial tonnage production without the need for extensive process re-optimization.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this NCL-based Semaglutide synthesis. These insights are derived directly from the experimental data and embodiments provided in the patent documentation, focusing on practical aspects of reaction optimization and impurity control. Understanding these nuances is essential for process chemists evaluating the feasibility of adopting this route for commercial production.
Q: How does this method overcome the solubility issues of hydrophobic Semaglutide fragments?
A: The patent utilizes high concentrations of Imidazole (2.5-5.0M) as an additive in the ligation buffer. This significantly enhances the solubility of the alkyl-side-chain-containing fragments (III and IV) while simultaneously catalyzing the native chemical ligation reaction without interfering with subsequent desulfurization.
Q: Why is the Glu-OAllyl protection strategy critical for thioester synthesis?
A: Peptide thioesters are unstable to the piperidine used in standard Fmoc deprotection. By anchoring the peptide to the resin via the side chain of Glutamic acid protected with an Allyl group, the main chain remains free for thioesterification after selective Allyl removal, bypassing the instability issue.
Q: What are the purification advantages of this fragment condensation approach?
A: Unlike stepwise SPPS which generates deletion peptides that are hard to separate, this method produces uncondensed fragments as the primary impurities. These fragments differ significantly in polarity from the final product, making HPLC purification much more efficient and allowing for the recycling of unreacted materials.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Semaglutide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic technologies to maintain competitiveness in the global pharmaceutical market. Our team of expert process chemists has extensively evaluated the NCL and desulfurization strategy outlined in CN114685645A and possesses the technical capability to implement this route at scale. We have extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to manufacturing plant is seamless and compliant with cGMP standards. Our rigorous QC labs and stringent purity specifications guarantee that every batch of Semaglutide intermediate or API meets the highest international quality benchmarks, providing our partners with the confidence needed for regulatory filings.
We invite procurement leaders and R&D directors to collaborate with us to leverage this innovative synthesis method for your supply chain. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team today to request specific COA data, route feasibility assessments, and a comprehensive proposal on how we can support your long-term Semaglutide sourcing strategy with reliability and excellence.