Optimizing Repaglinide Production: A Technical Analysis of the Novel Acyl Chloride Route
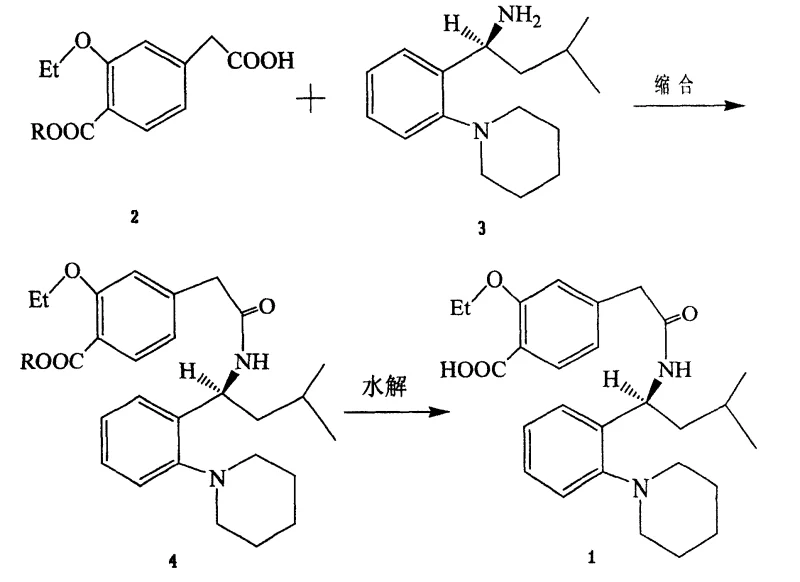
The pharmaceutical landscape for Type II diabetes treatment continues to evolve, with repaglinide standing out as a critical prandial glucose regulator. Patent CN100445275C introduces a robust and industrially viable synthetic pathway that addresses long-standing inefficiencies in producing this active pharmaceutical ingredient (API). Unlike traditional methods that rely on expensive coupling agents or generate stubborn impurities, this novel technique utilizes an acyl chloride intermediate strategy. By converting 4-carboxymethyl-3-ethoxybenzoate directly into an acid chloride before condensation, the process streamlines the workflow and enhances overall purity. This technical breakthrough is particularly relevant for reliable pharmaceutical intermediate suppliers aiming to optimize their manufacturing portfolios. The methodology not only promises improved reaction kinetics but also offers a clearer path to cost reduction in API manufacturing by utilizing readily available reagents such as thionyl chloride and common organic bases.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of repaglinide has been plagued by significant downstream processing challenges. Early literature, such as US5,312,924, described condensation reactions using carbonyldiimidazole (CDI) or N,N'-dicyclohexylcarbodiimide (DCC). While chemically feasible, these approaches introduce severe purification bottlenecks. Specifically, the use of DCC generates N,N'-dicyclohexylurea as a byproduct, which is notoriously difficult to remove completely without multiple recrystallizations, thereby driving up operational expenditures. Furthermore, CDI-mediated routes often suffer from mediocre yields, typically hovering between 50% and 55%, which is economically unsustainable for large-scale production. Alternative methods employing triphenylphosphine and carbon tetrachloride have shown slightly better yields but necessitate column chromatography for purification, a technique that is rarely feasible for multi-ton commercial manufacturing due to solvent consumption and throughput limitations.
The Novel Approach
The methodology outlined in CN100445275C represents a paradigm shift by bypassing these coupling agents entirely. Instead of activating the carboxylic acid with imidazoles or carbodiimides, the process converts the starting material into a highly reactive acid chloride intermediate. This activation step allows for a rapid and clean nucleophilic attack by the chiral amine component, S-(+)-1-(2-piperidinophenyl)-3-methylbutylamine. The result is a dramatic improvement in process efficiency, with the patent reporting a combined two-step yield reaching 80.9%. This approach eliminates the urea waste stream and removes the need for chromatographic purification, relying instead on simple crystallization and washing steps. For procurement teams, this translates to a more predictable supply chain and reduced waste disposal costs, making it a superior choice for the commercial scale-up of complex antidiabetic intermediates.
Mechanistic Insights into Acyl Chloride-Mediated Amidation
The core of this synthetic innovation lies in the efficient generation and utilization of the acid chloride species. In the first stage, the carboxylic acid precursor (Compound 2) reacts with chlorinating agents such as thionyl chloride, oxalyl chloride, or phosphorus pentachloride. This transformation replaces the hydroxyl group of the carboxylic acid with a chlorine atom, creating a potent electrophile. The reaction conditions are flexible, operating effectively across a temperature range of -10°C to 110°C depending on the specific chlorinating agent chosen. For instance, thionyl chloride refluxes efficiently at 40°C, while phosphorus pentachloride may require higher temperatures up to 110°C. This flexibility allows process chemists to tailor the reaction parameters to existing plant capabilities, ensuring that the activation step proceeds to completion with minimal side reactions.
Following activation, the introduction of the chiral amine (Compound 3) in the presence of a base facilitates the formation of the amide bond. The base, which can be an organic amine like triethylamine or an inorganic carbonate like potassium carbonate, serves a dual purpose: it neutralizes the hydrogen chloride gas evolved during the reaction and maintains the amine in its nucleophilic free-base form. Crucially, this mechanism avoids the racemization risks often associated with harsher coupling conditions, preserving the stereochemical integrity of the S-(+) configuration which is vital for biological activity. The subsequent hydrolysis step cleaves the ester protecting group under mild alkaline conditions (30°C to 70°C), yielding the final free acid. This gentle hydrolysis ensures that the sensitive amide linkage remains intact while effectively unmasking the pharmacophore, resulting in high-purity repaglinide suitable for final drug formulation.
How to Synthesize Repaglinide Efficiently
The synthesis protocol described in the patent provides a clear roadmap for laboratory and pilot-scale production. It emphasizes the importance of stoichiometric control, recommending a molar ratio of chlorinating agent to acid ranging from 0.5 to 4 equivalents to ensure complete conversion without excessive reagent waste. The detailed procedure involves dissolving the benzoate derivative in a solvent such as dichloromethane or toluene, followed by the controlled addition of the chlorinating agent. Once the acid chloride is formed, the chiral amine is introduced slowly to manage exotherms. The final hydrolysis is conducted in alcohol-water mixtures, allowing for easy pH adjustment to precipitate the product. For a comprehensive breakdown of the specific experimental parameters and safety considerations, please refer to the standardized guide below.
- Convert 4-carboxymethyl-3-ethoxybenzoate (Compound 2) into its corresponding acid chloride using thionyl chloride or oxalyl chloride in a solvent like dichloromethane.
- Condense the acid chloride intermediate with S-(+)-1-(2-piperidinophenyl)-3-methylbutylamine (Compound 3) in the presence of a base to form the protected ester (Compound 4).
- Hydrolyze the ester group of Compound 4 using an alkaline solution (e.g., NaOH or KOH) in a water-soluble solvent, followed by pH adjustment to isolate pure repaglinide.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of this acyl chloride route offers compelling economic benefits that extend beyond simple yield improvements. The elimination of specialized coupling reagents like CDI, which are significantly more expensive than commodity chemicals like thionyl chloride, directly impacts the bill of materials. Furthermore, the simplified workup procedure reduces the demand for extensive solvent volumes and energy-intensive purification steps. This efficiency gain is critical for maintaining competitive pricing in the global generic pharmaceutical market. By minimizing the number of unit operations and avoiding complex separation techniques, manufacturers can achieve faster batch turnover times, thereby enhancing overall equipment effectiveness and throughput capacity.
- Cost Reduction in Manufacturing: The substitution of high-cost coupling agents with inexpensive chlorinating reagents creates a substantial reduction in raw material expenses. Additionally, the avoidance of column chromatography and multiple recrystallizations lowers utility costs and solvent recovery burdens. The process relies on standard chemical transformations that utilize widely available industrial solvents, further insulating the supply chain from price volatility associated with specialty reagents. This streamlined approach ensures that production costs remain optimized without compromising the quality of the final API intermediate.
- Enhanced Supply Chain Reliability: The raw materials required for this synthesis, including thionyl chloride, triethylamine, and common esters, are commodity chemicals with robust global supply networks. This availability mitigates the risk of production delays caused by shortages of niche reagents. The simplicity of the reaction conditions also means that the process can be easily transferred between different manufacturing sites or scaled up using standard stainless steel reactors. Such flexibility ensures consistent supply continuity, a key metric for supply chain heads managing inventory for critical diabetes medications.
- Scalability and Environmental Compliance: The process generates fewer hazardous byproducts compared to DCC-mediated routes, specifically avoiding the accumulation of urea derivatives that require special disposal protocols. The use of aqueous workups and standard acid-base extractions aligns well with modern environmental, health, and safety (EHS) standards. Scalability is inherently supported by the homogeneous nature of the reaction phases and the absence of filtration-heavy purification steps. This makes the technology ideal for reducing lead time for high-purity pharmaceutical intermediates while adhering to strict regulatory guidelines regarding waste management and process safety.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this synthesis route. They are derived from the specific operational parameters and comparative data presented in the patent documentation. Understanding these nuances is essential for R&D teams evaluating technology transfer opportunities.
Q: What are the primary advantages of the acyl chloride method over CDI coupling for repaglinide?
A: The acyl chloride method avoids the formation of difficult-to-remove urea byproducts associated with DCC/CDI coupling, significantly simplifying purification and reducing production costs while achieving higher overall yields.
Q: What is the reported total yield for this synthesis route?
A: According to patent CN100445275C, the two-step reaction sequence achieves a total yield of approximately 80.9%, which is superior to previous methods reporting yields around 50-73%.
Q: Which solvents are compatible with the hydrolysis step?
A: The process utilizes water-soluble solvents such as methanol, ethanol, or acetone for the hydrolysis step, allowing for efficient reaction kinetics and straightforward product isolation via pH adjustment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Repaglinide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthetic routes in the competitive landscape of antidiabetic therapeutics. Our technical team has extensively analyzed the acyl chloride methodology and possesses the expertise to implement this process at an industrial scale. We boast extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of repaglinide intermediate meets the highest international standards for safety and efficacy.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific project requirements. By leveraging our manufacturing capabilities, you can access a Customized Cost-Saving Analysis tailored to your volume needs. We encourage you to contact us directly to request specific COA data and comprehensive route feasibility assessments. Let us collaborate to drive down costs and accelerate the delivery of life-saving medications to patients worldwide.