Advanced Control of Tofacitinib Intermediates for High-Purity API Manufacturing
Advanced Control of Tofacitinib Intermediates for High-Purity API Manufacturing
The pharmaceutical industry continuously faces challenges in maintaining stringent purity profiles for complex small molecule inhibitors, particularly for JAK inhibitors like Tofacitinib. Patent CN112592347A introduces a critical breakthrough in the synthesis of Tofacitinib intermediates, specifically addressing the formation and control of isopropyl-substituted impurities on the pyrrole nitrogen. This innovation is pivotal for manufacturers aiming to produce high-quality Active Pharmaceutical Ingredients (APIs) that meet rigorous global regulatory standards. By identifying the specific genesis of Related Substance I during the preparation of N-((3R,4R)-1-benzyl-4-methylpiperidin-3-yl)-N-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine, the patent provides a robust framework for upstream quality control. This technical advancement allows for the production of Tofacitinib with single impurity content consistently below 0.1%, a benchmark that is increasingly demanded by health authorities worldwide for chronic disease medications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for Tofacitinib often rely on a mature pathway involving condensation, deprotection, hydrogenation, and cyanoacetylation. However, a significant bottleneck in these conventional methods is the inadvertent formation of structurally similar impurities that are chemically stable and difficult to separate. Specifically, the alkylation of the pyrrole nitrogen with isopropyl groups creates a class of impurities that persist through multiple reaction steps. In standard processes without specific controls, these impurities can accumulate, leading to final API batches that fail to meet the strict specification of less than 0.1% for single unknown impurities. The removal of these isopropyl-substituted byproducts typically requires extensive and yield-destructive purification steps, such as repeated recrystallization or complex chromatographic separations, which drastically increases manufacturing costs and reduces overall process efficiency.
The Novel Approach
The novel approach detailed in the patent shifts the focus from downstream purification to upstream prevention. By rigorously controlling the content of Related Substance I in Intermediate 2 to be within 0.5% before proceeding to the next step, manufacturers can effectively prevent the propagation of these impurities into the final drug substance. This strategy leverages a deep understanding of the reaction mechanism, recognizing that the impurity is formed during the interaction of the intermediate with alkylating agents or solvents under specific conditions. Implementing this control point allows for a streamlined process where the final Tofacitinib product naturally meets purity specifications with minimal additional processing. This proactive quality-by-design (QbD) approach not only ensures compliance but also significantly enhances the economic viability of the manufacturing process by preserving yield.
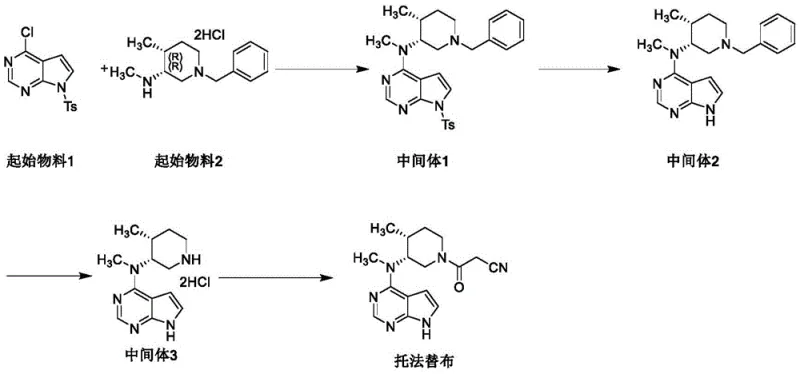
Mechanistic Insights into Isopropyl Substitution and Impurity Propagation
The formation of the critical impurity, identified as N-((3R,4R)-1-benzyl-4-methylpiperidin-3-yl)-7-isopropyl-N-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (Related Substance I), occurs through a nucleophilic substitution reaction on the pyrrole nitrogen. This reaction is facilitated by the presence of 2-halopropane or similar isopropyl sources under alkaline conditions. The mechanistic pathway involves the deprotonation of the pyrrole nitrogen, followed by an SN2 attack on the secondary carbon of the isopropyl halide. Once this isopropyl group is attached, it creates a steric and electronic environment that mimics the desired product, making separation via standard crystallization extremely challenging. Furthermore, this impurity is not inert; it survives the subsequent hydrogenation debenzylation step and the final cyanoacetylation, transforming into Related Substance III in the final API. This persistence underscores the necessity of intercepting the impurity at its source rather than attempting to remove it from the final complex molecule.
Understanding the kinetics of this side reaction is essential for process optimization. The patent data indicates that the formation of Related Substance I is influenced by reaction temperature, solvent choice, and the specific base used. For instance, the use of cesium carbonate in DMF at temperatures between 40-50°C can promote this alkylation if 2-halopropane is present as a contaminant or reagent. By mapping out this mechanistic vulnerability, process chemists can adjust reaction parameters to suppress the nucleophilicity of the pyrrole nitrogen or eliminate the source of the isopropyl group entirely. This level of mechanistic clarity empowers R&D teams to design more robust synthetic routes that are inherently resistant to the formation of these persistent byproducts, ensuring a cleaner reaction profile from the outset.
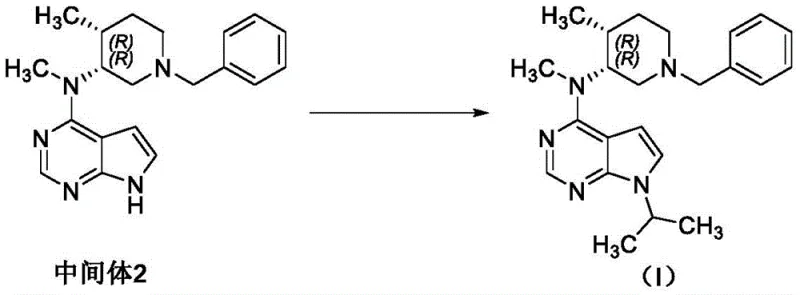
How to Synthesize Tofacitinib Efficiently
The efficient synthesis of high-purity Tofacitinib relies on a disciplined adherence to the controlled pathway outlined in the patent data. This involves a three-step sequence starting from the sulfonyl-protected precursor, moving through the critical Intermediate 2, and concluding with the final cyanoacetylation. The key to success lies in the meticulous monitoring of Intermediate 2 using high-performance liquid chromatography (HPLC) to ensure Related Substance I remains below the 0.5% threshold. Detailed standardized synthesis steps for implementing this impurity control strategy and achieving optimal yields are provided in the technical guide below.
- React N-((3R,4R)-1-benzyl-4-methylpiperidin-3-yl)-N-methyl-7-p-toluenesulfonyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine with NaOH in isopropanol to obtain Intermediate 2, ensuring related substance I content is controlled within 0.5%.
- Perform hydrogenation reduction on Intermediate 2 in the presence of dilute hydrochloric acid and Pd(OH)2/C catalyst to remove the benzyl group, yielding Intermediate 3.
- React Intermediate 3 with cyanoacetic acid N-hydroxysuccinimide ester and diisopropylethylamine to obtain high-purity Tofacitinib with single impurity content less than 0.1%.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the implementation of this impurity control technology translates directly into enhanced operational stability and cost efficiency. By adopting a synthesis route that minimizes the formation of difficult-to-remove impurities, manufacturers can reduce the reliance on expensive and time-consuming purification materials such as specialized chromatography resins or large volumes of recrystallization solvents. This reduction in downstream processing complexity leads to a more predictable production schedule, as the variability associated with struggling to meet purity specs is largely eliminated. Furthermore, the improved overall yield reported in the patent data suggests a more efficient utilization of raw materials, which is a critical factor in managing the cost of goods sold (COGS) for high-volume API production.
- Cost Reduction in Manufacturing: The ability to control impurities at the intermediate stage eliminates the need for extensive reprocessing or discarding of off-spec batches. By preventing the formation of Related Substance I, the process avoids the costly downstream steps usually required to scrub these impurities from the final API. This streamlined workflow reduces solvent consumption, energy usage, and labor hours associated with purification, resulting in substantial cost savings per kilogram of produced Tofacitinib. Additionally, the higher yield achieved through this method means that less starting material is required to produce the same amount of finished product, further driving down the unit cost of manufacturing.
- Enhanced Supply Chain Reliability: A robust manufacturing process that consistently meets purity specifications reduces the risk of batch failures and supply disruptions. When a production line is prone to generating hard-to-remove impurities, the lead time for releasing batches can extend significantly due to additional testing and rework. By integrating this control strategy, suppliers can offer more reliable delivery timelines, as the production cycle becomes more deterministic and less prone to unexpected quality deviations. This reliability is crucial for pharmaceutical companies managing just-in-time inventory systems and facing pressure to maintain continuous supply for patients dependent on chronic medication.
- Scalability and Environmental Compliance: The proposed method utilizes standard reagents and conditions that are amenable to large-scale commercial production, facilitating the transition from pilot plant to multi-ton manufacturing. Moreover, by reducing the volume of waste solvents and chemicals associated with repetitive purification steps, the process aligns better with green chemistry principles and environmental regulations. This reduced environmental footprint not only lowers waste disposal costs but also enhances the sustainability profile of the supply chain, a growing priority for global pharmaceutical purchasers evaluating their vendor networks.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the synthesis and quality control of Tofacitinib intermediates. These answers are derived from the specific experimental data and mechanistic insights provided in the patent literature, offering clarity on how impurity control impacts the final drug substance. Understanding these nuances is vital for technical teams evaluating the feasibility of adopting this improved synthetic route for commercial manufacturing.
Q: What is the primary source of difficult-to-remove impurities in Tofacitinib synthesis?
A: The primary source is the formation of isopropyl-substituted impurities on the pyrrole nitrogen (Related Substance I) during the preparation of Intermediate 2. If not controlled upstream, these propagate to the final API.
Q: Why is controlling Related Substance I in Intermediate 2 critical for final product quality?
A: Experiments demonstrate that if Related Substance I in Intermediate 2 exceeds 0.5%, it carries through subsequent hydrogenation and cyanoacetylation steps, resulting in final Tofacitinib impurities that are difficult to remove below the 0.1% threshold.
Q: How does this patented method improve overall process yield?
A: By strictly controlling the impurity profile at the Intermediate 2 stage, the need for extensive downstream purification is reduced, improving the total yield from approximately 60.1% to over 71.1% in comparative studies.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tofacitinib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the quality of the final API is inextricably linked to the purity of its precursors. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that complex impurity profiles like those described in CN112592347A are managed with precision. Our facility is equipped with rigorous QC labs capable of detecting trace impurities at the ppm level, guaranteeing that every batch of Tofacitinib intermediate we supply meets stringent purity specifications. We understand the critical nature of controlling Related Substance I and have integrated these advanced analytical and synthetic controls into our standard operating procedures to deliver consistent, high-quality materials.
We invite pharmaceutical partners to collaborate with us to leverage these process improvements for your supply chain. Our technical team is ready to provide a Customized Cost-Saving Analysis demonstrating how switching to our controlled intermediates can optimize your total manufacturing costs. Please contact our technical procurement team today to request specific COA data and route feasibility assessments tailored to your project requirements, ensuring a seamless path to market for your Tofacitinib formulations.