Optimizing Boron Neutron Capture Therapy Agents via Streamlined F-BPA Intermediate Synthesis
Optimizing Boron Neutron Capture Therapy Agents via Streamlined F-BPA Intermediate Synthesis
The rapidly evolving landscape of targeted cancer therapy has placed Boron Neutron Capture Therapy (BNCT) at the forefront of oncological innovation, driving an urgent demand for high-purity radiopharmaceutical precursors. Patent CN115043864A introduces a groundbreaking methodology for synthesizing intermediates crucial for the production of 2-fluoro-4-dihydroxyboron-L-phenylalanine, commonly known as F-BPA. This technical disclosure addresses critical bottlenecks in existing nucleophilic substitution methods, which traditionally suffer from protracted reaction times and suboptimal yields due to excessive separation steps post-fluorination. By streamlining the synthetic pathway to require only a single hydrolysis reaction after the fluorination step, this invention not only accelerates the total synthesis time but also markedly enhances the radiochemical yield and specific activity of the final product. For industry stakeholders, this represents a pivotal shift towards more efficient, cost-effective, and clinically viable production of BNCT agents.
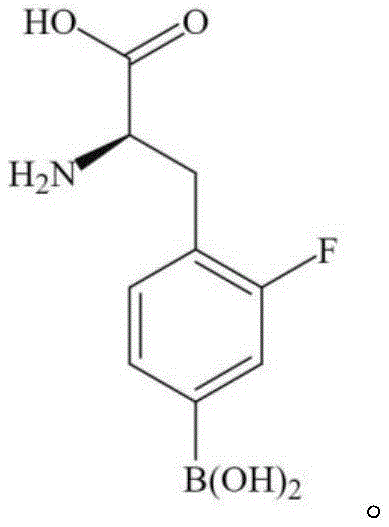
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of F-BPA and its radiolabeled counterpart 18F-BPA has been plagued by inefficient multi-step protocols that severely impact both throughput and product viability. Conventional nucleophilic substitution methods often necessitate complex purification sequences immediately following the introduction of the fluorine atom, leading to significant losses in radioactivity due to the short half-life of fluorine-18. These legacy processes typically involve harsh reaction conditions that can compromise the integrity of the sensitive boron moiety, resulting in impurity profiles that are difficult to manage without extensive chromatographic intervention. Furthermore, the reliance on multiple isolation steps increases the operational complexity and solvent consumption, creating substantial barriers to scaling these reactions for commercial supply chains. The cumulative effect of these inefficiencies is a final product with lower specific activity and radiochemical purity, which fails to meet the stringent requirements for human clinical imaging and therapy.
The Novel Approach
In stark contrast, the methodology outlined in patent CN115043864A offers a refined synthetic trajectory that elegantly bypasses these traditional hurdles through strategic intermediate design. The core innovation lies in the construction of a robust precursor, specifically the third compound depicted in the patent, which serves as a stable platform for subsequent fluorination. 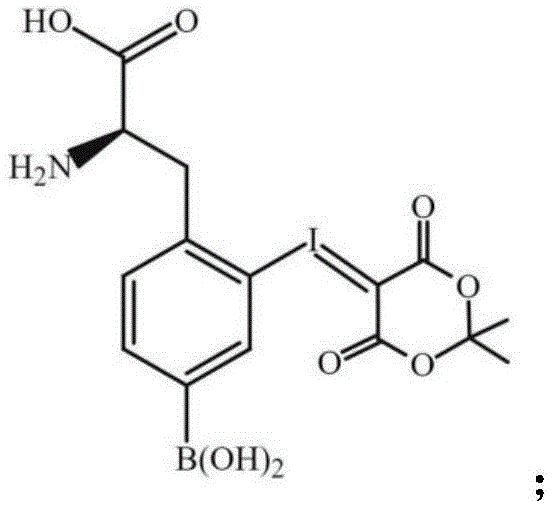 By pre-installing the necessary structural features before the critical fluorination event, the process eliminates the need for post-fluorination structural manipulations other than a simple hydrolysis. This approach drastically reduces the number of unit operations required, thereby minimizing the time window between radionuclide production and final drug formulation. The result is a synthesis protocol that is not only chemically superior in terms of yield but also operationally streamlined, offering a clear pathway for reliable pharmaceutical intermediate supplier networks to adopt.
By pre-installing the necessary structural features before the critical fluorination event, the process eliminates the need for post-fluorination structural manipulations other than a simple hydrolysis. This approach drastically reduces the number of unit operations required, thereby minimizing the time window between radionuclide production and final drug formulation. The result is a synthesis protocol that is not only chemically superior in terms of yield but also operationally streamlined, offering a clear pathway for reliable pharmaceutical intermediate supplier networks to adopt.
Mechanistic Insights into Iodination and Nucleophilic Fluorination
The chemical elegance of this process is rooted in a precise sequence of electrophilic aromatic substitution followed by nucleophilic displacement, orchestrated to maximize regioselectivity and conversion efficiency. The initial phase involves the generation of iodine monochloride (ICl) in situ from iodide ions and hypochlorite under acidic conditions, which then reacts with the starting phenylalanine derivative to install an iodine leaving group at the ortho position relative to the side chain. This iodinated intermediate is subsequently oxidized using m-chloroperoxybenzoic acid (mCPBA) to activate the aromatic ring for coupling with isopropylidene malonate, forming a stable cyclic structure that protects the boron functionality during subsequent harsh conditions. The strategic use of Meldrum's acid derivatives ensures that the boron atom remains intact and unreactive until the final deprotection stage, a critical factor in maintaining the therapeutic efficacy of the BNCT agent.
Following the construction of the carbon skeleton, the amine group is protected using di-tert-butyl dicarbonate to prevent unwanted side reactions during the high-temperature fluorination step. 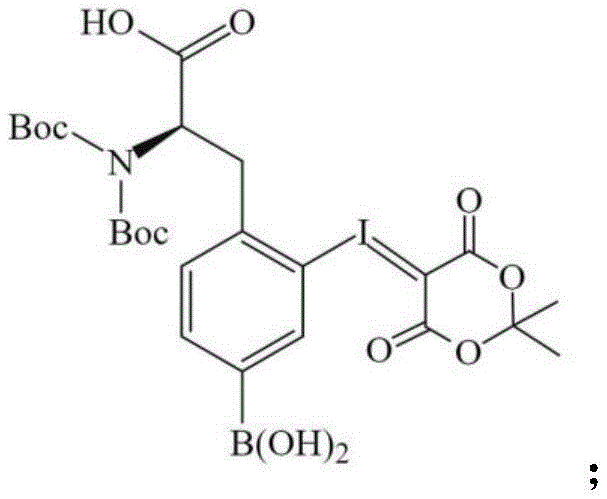 The pivotal fluorination event utilizes potassium fluoride complexed with Kryptofix 2.2.2 in anhydrous DMSO, facilitating a potent nucleophilic aromatic substitution that displaces the iodine atom with high specificity. This step is conducted at elevated temperatures ranging from 120°C to 150°C, conditions that are rigorously controlled to ensure complete conversion while preventing thermal degradation of the sensitive boronate ester. The mechanistic robustness of this sequence allows for the consistent production of the fifth compound with high radiochemical purity, setting the stage for a straightforward final hydrolysis that reveals the active F-BPA molecule without generating significant byproduct contamination.
The pivotal fluorination event utilizes potassium fluoride complexed with Kryptofix 2.2.2 in anhydrous DMSO, facilitating a potent nucleophilic aromatic substitution that displaces the iodine atom with high specificity. This step is conducted at elevated temperatures ranging from 120°C to 150°C, conditions that are rigorously controlled to ensure complete conversion while preventing thermal degradation of the sensitive boronate ester. The mechanistic robustness of this sequence allows for the consistent production of the fifth compound with high radiochemical purity, setting the stage for a straightforward final hydrolysis that reveals the active F-BPA molecule without generating significant byproduct contamination.
How to Synthesize F-BPA Intermediates Efficiently
Executing this synthesis requires strict adherence to the reaction parameters defined in the patent to ensure optimal yield and purity profiles suitable for clinical applications. The process begins with the careful preparation of the iodinating mixture, followed by the sequential addition of reagents to build the complex molecular architecture step-by-step. Operators must maintain anhydrous conditions during the fluorination phase to prevent hydrolysis of the reactive fluoride species, while the final hydrolysis step requires precise acid concentration control to cleave the protecting groups without damaging the boron-carbon bond. Detailed standardized synthesis steps see the guide below for specific stoichiometric ratios and temperature ramps.
- React the starting phenylalanine derivative with iodide ions and hypochlorite under acidic conditions to generate the iodinated intermediate.
- Couple the iodinated intermediate with isopropylidene malonate (Meldrum's acid) using mCPBA oxidation to form the protected boron-containing scaffold.
- Protect the amine group with Boc anhydride, followed by nucleophilic substitution with fluoride ions using Kryptofix 2.2.2 to yield the fluorinated precursor.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route translates directly into tangible operational efficiencies and risk mitigation strategies. By reducing the total number of synthetic steps and eliminating complex purification stages post-fluorination, manufacturers can significantly lower the consumption of high-grade solvents and specialized chromatography media. This simplification of the workflow reduces the dependency on scarce skilled labor for intricate separation tasks, allowing facilities to increase batch frequency and overall throughput without proportional increases in capital expenditure. Furthermore, the improved stability of the intermediates allows for more flexible inventory management, reducing the pressure on just-in-time delivery models that often plague radiopharmaceutical supply chains.
- Cost Reduction in Manufacturing: The elimination of multiple separation steps and the use of commercially available reagents like Meldrum's acid and standard Boc anhydride drive down the raw material costs significantly. By avoiding the need for expensive transition metal catalysts or exotic ligands often required in cross-coupling alternatives, the process achieves substantial cost savings in reagent procurement. Additionally, the higher overall yield means less starting material is wasted, directly improving the cost-per-gram metric for the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The robustness of the synthetic route ensures consistent output quality, which is critical for maintaining regulatory compliance and avoiding costly batch failures. Since the method relies on stable intermediates that can be characterized and stored prior to the final radiolabeling step, it decouples the complex organic synthesis from the time-sensitive radioactive labeling, providing a buffer against supply disruptions. This modularity allows suppliers to stockpile key precursors, ensuring continuity of supply even when cyclotron availability or logistics face unexpected challenges.
- Scalability and Environmental Compliance: The process utilizes common organic solvents such as dichloromethane and DMSO, which are well-understood in terms of waste treatment and recycling, simplifying environmental compliance protocols. The reduction in reaction steps inherently lowers the volume of chemical waste generated per kilogram of product, aligning with green chemistry principles and reducing disposal costs. This scalability makes the technology ideal for commercial scale-up of complex amino acid derivatives, enabling manufacturers to meet growing global demand for BNCT therapies without expanding their physical footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and benefits of this synthesis technology, derived directly from the experimental data and claims within the patent documentation. Understanding these nuances is essential for R&D teams evaluating the feasibility of integrating this route into their existing production pipelines. The answers reflect the specific advantages related to yield, purity, and operational simplicity that distinguish this method from prior art.
Q: What is the primary advantage of the synthesis method in CN115043864A?
A: The method significantly shortens the synthesis time after fluorination substitution by requiring only one hydrolysis step to obtain the final F-BPA product, thereby improving overall radiochemical yield.
Q: What radiochemical purity can be achieved with this process?
A: The process achieves a radiochemical purity of approximately 98% for 18F-BPA, as measured by thin-layer chromatography, ensuring high quality for clinical diagnostic applications.
Q: Is this synthesis route suitable for large-scale manufacturing?
A: Yes, the route utilizes standard organic solvents like dichloromethane and DMSO and avoids exotic catalysts, making it highly adaptable for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable F-BPA Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthesis methods described in CN115043864A for the future of Boron Neutron Capture Therapy. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to market-ready supply is seamless and secure. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications for radiopharmaceutical intermediates, guaranteeing that every batch of F-BPA or its precursors meets the highest international standards for safety and efficacy.
We invite you to collaborate with our technical procurement team to explore how this advanced synthesis route can optimize your specific manufacturing requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of adopting this streamlined process for your supply chain. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments tailored to your project timelines, ensuring that your development programs proceed with the reliability and speed they deserve.