Advanced Nucleophilic Fluorination Strategy for High-Purity F-BPA Production
The pharmaceutical landscape for Boron Neutron Capture Therapy (BNCT) and Positron Emission Tomography (PET) imaging is undergoing a significant transformation driven by the innovations detailed in patent CN108299482B. This pivotal intellectual property introduces a robust nucleophilic fluorination synthesis method for F-BPA (4-borono-2-[18F]fluoro-L-phenylalanine), addressing critical limitations inherent in traditional electrophilic labeling techniques. As a leading entity in the fine chemical sector, we recognize that the shift from electrophilic to nucleophilic pathways represents not merely a procedural adjustment but a fundamental upgrade in radiochemical purity and specific activity. The patent outlines a sophisticated five-step sequence that begins with simple boron-containing precursors and culminates in the highly sought-after radiopharmaceutical intermediate. For research directors and procurement specialists alike, understanding the nuances of this technology is essential for securing a reliable radiopharmaceutical intermediate supplier capable of meeting the stringent demands of modern oncology diagnostics and therapy.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the preparation of 18F-BPA has relied heavily on electrophilic fluorination methods utilizing [18F]F2 gas. While conceptually straightforward, this approach suffers from severe drawbacks that hinder its clinical and commercial viability. The primary issue lies in the low specific activity of the resulting product, typically ranging between 35-60 MBq/μmol, due to the inevitable introduction of stable fluorine carriers during the gas target processing. Furthermore, the handling of elemental fluorine gas presents substantial safety hazards, requiring specialized, corrosion-resistant equipment that drastically inflates capital expenditure. The operational complexity of managing gas-phase radiochemistry also introduces significant bottlenecks in production throughput, making it difficult to scale for widespread clinical application. These factors collectively limit the efficacy of BNCT treatment planning, as lower specific activity can compromise the precision of tumor localization and dosimetry calculations.
The Novel Approach
In stark contrast, the methodology disclosed in CN108299482B leverages nucleophilic substitution using no-carrier-added [18F]fluoride ions. This paradigm shift eliminates the need for hazardous F2 gas entirely, replacing it with safer, solution-phase chemistry that is compatible with standard automated synthesis modules. By utilizing a tailored precursor featuring a suitable leaving group, the process achieves high radiochemical yields without diluting the specific activity with stable isotopes. The result is a product with superior imaging characteristics, capable of providing clearer metabolic maps of glioma and melanoma tissues. This novel approach not only enhances the diagnostic quality but also streamlines the manufacturing workflow, offering a pathway for cost reduction in PET tracer manufacturing that is both economically and environmentally sustainable for large-scale operations.
Mechanistic Insights into Nucleophilic Aromatic Substitution and Chiral Catalysis
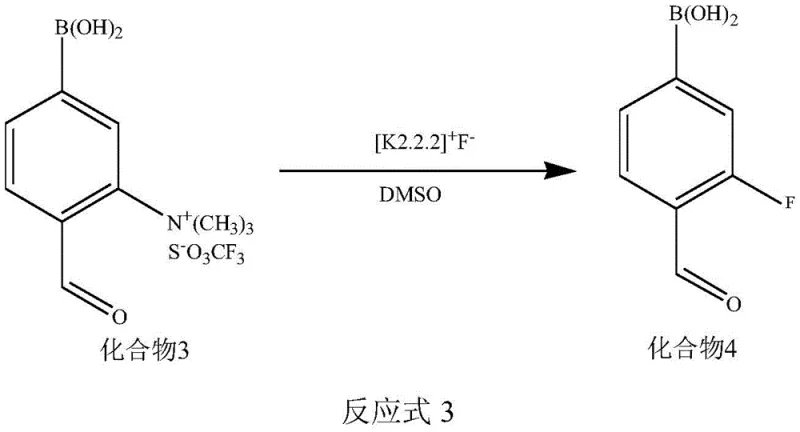
The core of this synthetic innovation lies in the precise execution of nucleophilic aromatic substitution followed by enantioselective alkylation. The process initiates with the conversion of a fluoro-aldehyde precursor into a dimethylamino derivative, which is subsequently quaternized to form a trimethylammonium salt. This salt serves as an excellent leaving group for the subsequent nucleophilic attack by the fluoride ion, facilitated by the Kryptofix 2.2.2 (K2.2.2) cryptand system. The cryptand complexes the potassium cation, effectively "naked" the fluoride anion and enhancing its nucleophilicity in polar aprotic solvents like DMSO. This activation allows the fluorine atom to displace the trimethylammonium group under relatively mild thermal conditions, preserving the integrity of the sensitive boronic acid moiety which is crucial for the biological targeting of the final drug.
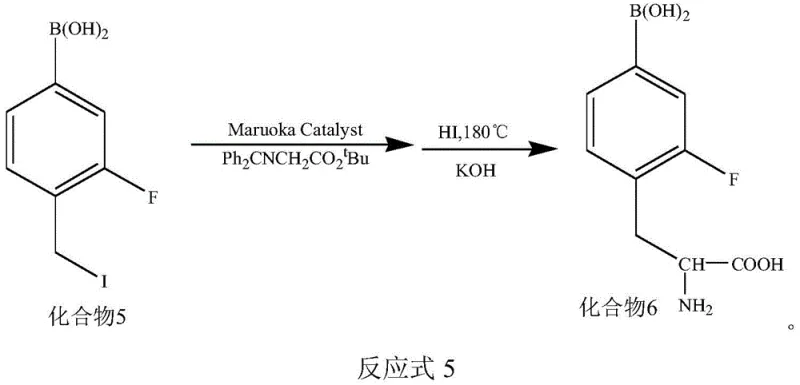
Following the fluorination event, the synthesis proceeds through a reductive iodination and concludes with a Maruoka-catalyzed asymmetric alkylation. This final step is critical for establishing the correct stereochemistry at the alpha-carbon of the amino acid backbone. The Maruoka chiral phase transfer catalyst creates a rigid chiral environment that directs the incoming glycine equivalent to attack the alkyl iodide from a specific face, ensuring the exclusive formation of the L-enantiomer. Impurity control is rigorously maintained throughout the sequence via solid-phase extraction techniques using Sep-Pak C18 and Silican columns, which effectively remove unreacted precursors, side products, and metal residues. This meticulous attention to purification ensures that the final high-purity OLED material or pharmaceutical intermediate meets the rigorous standards required for human injection.
How to Synthesize F-BPA Efficiently
Implementing this synthesis requires a disciplined approach to reaction conditions and purification protocols to maximize yield and radiochemical purity. The process is designed to be modular, allowing for the isolation of stable intermediates which can be quality-controlled before proceeding to the radioactive labeling step. This modularity is particularly advantageous for commercial scale-up of complex radiopharmaceuticals, as it decouples the non-radioactive precursor synthesis from the short-half-life radiolabeling event. Operators must pay close attention to the moisture content during the fluorination step, as water can compete with fluoride and reduce labeling efficiency. Furthermore, the temperature control during the Maruoka catalysis step is vital to prevent racemization. For a detailed breakdown of the specific molar ratios, solvent systems, and reaction times validated in the patent examples, please refer to the standardized protocol below.
- Synthesize Compound 2 by reacting Compound 1 with dimethylammonium hydrochloride and potassium carbonate in DMSO/water under reflux.
- Convert Compound 2 to Compound 3 using methyl-trifluoromethanesulfonate in dichloromethane under inert gas protection.
- Perform nucleophilic fluorination on Compound 3 using K2.2.2/KF complex in DMSO at elevated temperatures to yield Compound 4.
- Reduce the aldehyde group in Compound 4 using NaBH4 and convert to iodide Compound 5 using HI solution on a solid phase column.
- Couple Compound 5 with N-(diphenylmethyl)-glycine tert-butyl ester using Maruoka catalyst, followed by hydrolysis to obtain final F-BPA.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this nucleophilic synthesis route offers tangible strategic benefits beyond mere technical superiority. The elimination of fluorine gas handling infrastructure translates directly into reduced facility maintenance costs and lower regulatory compliance burdens associated with hazardous gas storage. Moreover, the stability of the non-radioactive precursors allows for bulk purchasing and long-term inventory management, mitigating the risks of supply chain disruptions that often plague just-in-time radiochemistry models. The use of common organic solvents and standard laboratory glassware further democratizes the production capability, enabling a broader network of qualified manufacturers to enter the market. This increased competition and accessibility drive down overall procurement costs while enhancing supply security for critical cancer therapeutics.
- Cost Reduction in Manufacturing: The removal of expensive gas-target systems and the associated specialized piping significantly lowers the barrier to entry for production facilities. By relying on solution-phase chemistry, manufacturers can utilize existing organic synthesis suites without major retrofitting. Additionally, the higher specific activity of the product means that less precursor mass is required to achieve the same diagnostic signal, effectively reducing the cost of goods sold per patient dose. The streamlined purification process using solid-phase columns also minimizes solvent consumption and waste disposal costs, contributing to a leaner and more cost-effective manufacturing profile.
- Enhanced Supply Chain Reliability: The precursors described in this patent, such as the trimethylammonium salts, are chemically stable and can be synthesized in large batches well in advance of the radiolabeling event. This decoupling allows suppliers to build robust safety stocks, ensuring continuity of supply even in the face of raw material fluctuations. Unlike electrophilic methods that depend on the immediate availability of cyclotron-produced gas, this nucleophilic route offers greater flexibility in scheduling and logistics. It enables a more resilient supply chain capable of supporting multi-center clinical trials and widespread commercial distribution without the bottleneck of gas transport.
- Scalability and Environmental Compliance: The synthetic route avoids the generation of corrosive fluorine waste streams, simplifying effluent treatment and aligning with green chemistry principles. The reactions are conducted in closed vessels with standard workup procedures like extraction and crystallization, which are inherently easier to scale from gram to kilogram quantities compared to gas-phase reactions. This scalability ensures that as demand for BNCT drugs grows, production capacity can be expanded rapidly without encountering the engineering challenges associated with scaling hazardous gas chemistry. The environmental footprint is further reduced by the efficient use of reagents and the potential for solvent recycling.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of adopting this synthesis method, we have compiled answers to common inquiries regarding the process parameters and product specifications. These insights are derived directly from the experimental data and claims within the patent documentation, providing a factual basis for decision-making. Understanding these details is crucial for integrating this technology into existing GMP workflows and ensuring that the final product meets all regulatory requirements for clinical use. We encourage stakeholders to review these points carefully to appreciate the full scope of the technical advantages offered by this nucleophilic approach.
Q: What is the primary advantage of this nucleophilic method over electrophilic fluorination?
A: The nucleophilic method avoids the use of carrier-added [18F]F2 gas, resulting in products with significantly higher specific activity and no stable fluorine carrier, which is critical for high-quality PET imaging.
Q: How does the Maruoka catalyst contribute to the synthesis?
A: The Maruoka chiral phase transfer catalyst facilitates the asymmetric alkylation in the final step, ensuring the production of the biologically active L-enantiomer of BPA with high stereochemical purity.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process utilizes standard organic synthesis techniques such as reflux, extraction, and solid-phase purification, avoiding the need for specialized gas-target infrastructure required by electrophilic methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable F-BPA Supplier
At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, positioning us as an ideal partner for bringing this advanced F-BPA technology to market. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of radiopharmaceutical intermediate meets the highest global standards. We understand the critical nature of supply continuity in the healthcare sector and have optimized our operations to deliver consistent quality and reliability. By leveraging our expertise in chiral catalysis and nucleophilic fluorination, we can help you accelerate your development timelines and secure a competitive edge in the oncology market.
We invite you to engage with our technical procurement team to discuss how we can tailor our manufacturing capabilities to your specific needs. Whether you require a Customized Cost-Saving Analysis for transitioning from electrophilic to nucleophilic methods or need specific COA data to validate our process against your internal benchmarks, we are ready to provide comprehensive support. Let us collaborate to optimize your route feasibility assessments and ensure a seamless supply of high-quality intermediates for your next-generation PET imaging and BNCT applications.