Optimizing BNCT Agent Production: A Technical Analysis of the Novel F-BPA Synthesis Route
Optimizing BNCT Agent Production: A Technical Analysis of the Novel F-BPA Synthesis Route
The landscape of Boron Neutron Capture Therapy (BNCT) and Positron Emission Tomography (PET) imaging is constantly evolving, driven by the critical need for high-purity radiopharmaceutical precursors. A pivotal development in this sector is documented in patent CN115010739A, which discloses a highly efficient method for synthesizing F-BPA (2-fluoro-4-borono-L-phenylalanine). This compound serves as a crucial cold standard and precursor for 18F-BPA, a radiotracer used to monitor drug distribution in tumor tissues. The technical breakthrough lies in the streamlining of the synthetic pathway, specifically addressing the bottleneck of post-fluorination processing. By optimizing the sequence of protection, substitution, and deprotection, this methodology offers a robust solution for producing high-purity intermediates essential for clinical applications.
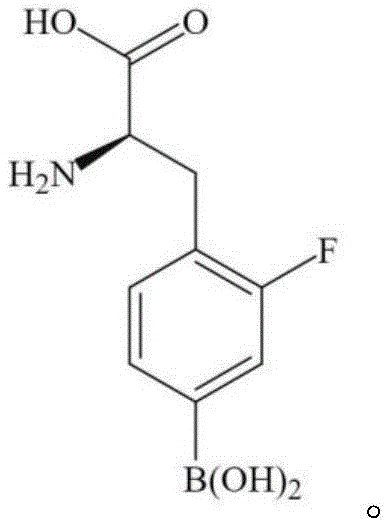
For R&D directors and procurement specialists, the implications of this patent extend beyond mere academic interest. The ability to synthesize F-BPA with improved yield and reduced reaction time directly correlates to enhanced supply chain stability and cost efficiency. Traditional routes often suffer from cumulative yield losses due to excessive purification steps required after the delicate fluorination reaction. This new approach mitigates those risks by ensuring that once the fluorine atom is successfully incorporated into the aromatic ring, the molecule is仅需 one hydrolysis step away from the final active pharmaceutical ingredient (API) intermediate. This structural elegance minimizes exposure to harsh conditions that could degrade the sensitive boronic acid moiety, thereby preserving the integrity of the final product.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fluorinated amino acid derivatives like F-BPA has been plagued by inefficiencies inherent in multi-step organic synthesis. Conventional nucleophilic substitution methods typically involve introducing the fluorine atom at a stage where the molecule still possesses multiple reactive functional groups that require extensive protection. Following the fluorination event, older methodologies often necessitated a cascade of deprotection reactions, each requiring separate work-up procedures, solvent exchanges, and purification cycles. This not only extends the total synthesis time—a critical factor when dealing with radioactive isotopes—but also significantly diminishes the overall yield. Each additional step introduces a potential point of failure, increasing the likelihood of impurity formation and complicating the final purification profile. Furthermore, the prolonged exposure of the boronic acid group to various chemical environments in these lengthy sequences can lead to protodeboronation or oxidation, resulting in substantial material loss and increased production costs.
The Novel Approach
The methodology outlined in patent CN115010739A represents a paradigm shift by decoupling the complexity of the synthesis from the final fluorination step. The core innovation is the design of a precursor (Compound 4) that is pre-optimized for nucleophilic attack. By strategically employing a Meldrum's acid derivative and a Boc-protecting group prior to fluorination, the molecule is rendered stable enough to withstand the rigorous conditions required for aromatic substitution (120-150°C in DMSO). Crucially, the patent demonstrates that after the fluorine is introduced to form Compound 5, the remaining protecting groups can be cleaved simultaneously in a single hydrolysis step. This consolidation of the final deprotection phase eliminates the need for intermediate isolations and multiple reaction vessels. For a reliable radiopharmaceutical intermediate supplier, this translates to a process that is not only faster but inherently more robust, reducing the operational burden and minimizing the generation of chemical waste associated with repetitive purification.
Mechanistic Insights into Nucleophilic Aromatic Substitution and Protection Strategies
To fully appreciate the technical sophistication of this route, one must examine the specific chemical transformations employed. The synthesis begins with the electrophilic iodination of the starting borono-phenylalanine derivative. This step is critical as it installs the leaving group (iodine) necessary for the subsequent nucleophilic aromatic substitution (SnAr). The reaction utilizes an iodide source and an oxidant in an acidic medium to generate the active iodinating species in situ, ensuring high regioselectivity for the position ortho to the boronic acid group. Following this, the carboxylic acid functionality is masked using isopropylidene malonate (Meldrum's acid). This forms a cyclic structure that protects the acid while potentially activating the ring system for the upcoming substitution. The amine group is then capped with a tert-butoxycarbonyl (Boc) group using di-tert-butyl dicarbonate. This dual-protection strategy is vital; without it, the nucleophilic amine or the acidic proton of the carboxyl group could interfere with the fluorination reagents or undergo degradation under the high-temperature conditions required for the SnAr reaction.
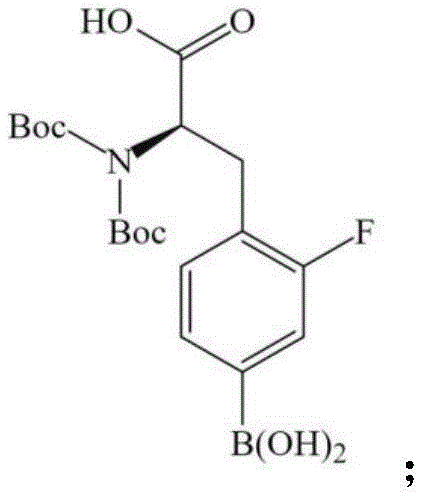
The centerpiece of the mechanism is the nucleophilic fluorination step, where the iodo-intermediate reacts with fluoride ions. In this specific embodiment, potassium fluoride (KF) is complexed with Kryptofix 2.2.2 (K2.2.2) in anhydrous dimethyl sulfoxide (DMSO). The cryptand acts as a phase-transfer catalyst, sequestering the potassium cation and rendering the fluoride anion "naked" and highly nucleophilic. This allows the fluoride to displace the iodine atom on the electron-deficient aromatic ring efficiently. The patent specifies reaction temperatures between 120°C and 150°C, conditions that would typically destroy unprotected amino acids. However, thanks to the robust Boc and Meldrum's acid protections, the scaffold remains intact. The final step involves acid-catalyzed hydrolysis, likely using hydrochloric acid, which cleaves both the Boc group and the Meldrum's acid derivative simultaneously. This elegant "one-pot" deprotection reveals the free amine and carboxylic acid, yielding the target F-BPA with high radiochemical purity, reported in the patent examples to be approximately 98%.
How to Synthesize 2-fluoro-4-borono-L-phenylalanine Efficiently
The synthesis of F-BPA described in this patent offers a reproducible framework for manufacturing high-quality intermediates. The process relies on well-established organic transformations but arranges them in a novel sequence to maximize efficiency. Operators should pay close attention to the anhydrous conditions required during the fluorination step, as moisture can severely inhibit the nucleophilicity of the fluoride ion. Additionally, the purification of the intermediates, particularly the transition from the iodo-compound to the protected precursor, sets the stage for the success of the final fluorination. Detailed standardized operating procedures regarding reagent stoichiometry, temperature ramping, and work-up protocols are essential to replicate the yields described in the intellectual property. For comprehensive technical execution, please refer to the specific experimental guidelines provided below.
- Perform electrophilic iodination on the starting borono-phenylalanine derivative using an iodide source and oxidant in acidic media to generate the iodo-intermediate.
- Condense the iodo-intermediate with isopropylidene malonate (Meldrum's acid) under basic conditions to form the protected cyclic intermediate.
- Protect the amine group using di-tert-butyl dicarbonate (Boc2O) to prevent side reactions during the subsequent harsh fluorination step.
- Execute nucleophilic aromatic substitution using potassium fluoride (KF) complexed with Kryptofix 2.2.2 in anhydrous DMSO to introduce the fluorine atom.
- Conduct a final acid-catalyzed hydrolysis to remove protecting groups and yield the pure F-BPA product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthesis route offers tangible benefits for procurement managers and supply chain heads looking to optimize their sourcing strategies for BNCT agents. The primary value driver is the drastic simplification of the manufacturing workflow. By reducing the number of unit operations required post-fluorination, the process inherently lowers the consumption of solvents, reagents, and labor hours. This reduction in processing intensity directly contributes to cost reduction in PET tracer manufacturing, allowing suppliers to offer more competitive pricing without compromising on quality. Furthermore, the shortened synthesis time is particularly advantageous for the production of radioactive batches, where the half-life of the isotope dictates the maximum allowable processing window. A faster route means higher specific activity at the time of administration, which is a key quality attribute for radiopharmaceuticals.
- Cost Reduction in Manufacturing: The elimination of multiple separation and purification steps after the fluorination reaction leads to significant operational savings. In traditional multi-step syntheses, each isolation event incurs costs related to chromatography media, solvent disposal, and equipment occupancy time. By consolidating the final deprotection into a single hydrolysis step, this method minimizes the physical footprint of the production line and reduces the volume of hazardous waste generated. Qualitatively, this implies a leaner manufacturing process where the cost of goods sold (COGS) is optimized through efficiency rather than cheap raw materials. The higher overall yield also means that less expensive starting material is wasted, further enhancing the economic viability of large-scale production runs.
- Enhanced Supply Chain Reliability: Supply chain continuity is often threatened by complex processes that are prone to failure at multiple stages. This streamlined route enhances reliability by reducing the number of critical control points where a batch could be rejected. The use of stable, storable intermediates (such as the Boc-protected precursor) allows manufacturers to produce and stockpile key building blocks in advance, decoupling the supply of the final API from the immediate availability of all reagents. This buffering capability ensures that demand spikes for F-BPA can be met with shorter lead times. Additionally, the reliance on commodity chemicals like KF, DMSO, and Boc2O, rather than exotic catalysts, mitigates the risk of supply disruptions for critical reagents, ensuring a steady flow of materials for continuous manufacturing.
- Scalability and Environmental Compliance: The scalability of this process is supported by its use of standard reaction conditions and widely available solvents. Unlike methods requiring cryogenic temperatures or ultra-high vacuum, this route operates at manageable temperatures and pressures, facilitating easy scale-up from gram to kilogram quantities. From an environmental standpoint, the reduction in solvent usage and waste generation aligns with green chemistry principles. Fewer purification steps mean less silica gel and eluent waste, simplifying the compliance burden for waste treatment facilities. This environmental efficiency not only reduces disposal costs but also positions the manufacturer favorably in markets with stringent environmental regulations, ensuring long-term operational sustainability.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of F-BPA synthesized via this patented method. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the feasibility and advantages of this specific chemical route. Understanding these details is crucial for stakeholders evaluating the integration of this intermediate into their broader radiopharmaceutical supply chains.
Q: What is the primary advantage of this F-BPA synthesis method over conventional nucleophilic substitution routes?
A: The primary advantage is the significant reduction in post-fluorination processing steps. Conventional methods often require multiple synthesis and separation steps after introducing the fluorine atom, leading to long reaction times and lower yields. This patented method achieves the final product through only one hydrolysis reaction after fluorination, drastically shortening the total synthesis time and improving the overall radiochemical yield, which is critical for short-half-life isotopes like 18F.
Q: How does the use of Meldrum's acid and Boc protection enhance the process reliability?
A: The strategic use of Meldrum's acid (isopropylidene malonate) and tert-butoxycarbonyl (Boc) groups serves to protect sensitive functional groups on the phenylalanine backbone. This protection prevents unwanted side reactions during the high-temperature nucleophilic fluorination step (120-150°C). By stabilizing the intermediate structure, the process ensures higher purity of the fluoro-intermediate, which simplifies the final purification and reduces the formation of difficult-to-remove impurities.
Q: Is this synthesis route scalable for commercial production of non-radioactive F-BPA standards?
A: Yes, the route is highly scalable. The reaction conditions utilize standard organic solvents like dichloromethane and DMSO, and common reagents such as potassium fluoride and sodium bicarbonate. The isolation methods involve standard techniques like extraction, recrystallization, and column chromatography (silica or C18), which are easily transferable from laboratory scale to multi-kilogram commercial manufacturing without requiring specialized exotic equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-fluoro-4-borono-L-phenylalanine Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful deployment of BNCT therapies relies on the consistent availability of high-quality precursors like F-BPA. Our technical team has extensively analyzed the route described in CN115010739A and possesses the expertise to implement this streamlined synthesis at an industrial scale. We understand that transitioning from a patent concept to commercial reality requires rigorous process validation. Therefore, we leverage our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production to ensure that every batch meets stringent purity specifications. Our rigorous QC labs are equipped to verify the radiochemical purity and specific activity of the final product, ensuring it performs reliably in clinical imaging and therapeutic monitoring applications.
We invite pharmaceutical partners and research institutions to collaborate with us to secure a stable supply of this critical intermediate. By optimizing the synthesis parameters further, we can tailor the production process to meet your specific volume requirements and timeline constraints. Please contact our technical procurement team to request a Customized Cost-Saving Analysis for your project. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your R&D and commercial goals effectively.