Advanced Synthesis of Decitabine Intermediates: Overcoming Stereoselectivity Challenges for Commercial Scale
Advanced Synthesis of Decitabine Intermediates: Overcoming Stereoselectivity Challenges for Commercial Scale
The pharmaceutical landscape for oncology treatments continues to evolve, with nucleoside analogues like decitabine playing a pivotal role in managing myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). However, the commercial viability of these potent therapeutics often hinges on the efficiency of their synthetic routes, particularly regarding stereoselectivity and purification scalability. Patent CN111377989B introduces a transformative preparation method for a key decitabine intermediate that addresses long-standing industry bottlenecks. By leveraging a specific 2-deoxy-D-ribose derivative coupled with an activated 5-azacytosine base, this technology achieves a remarkable enhancement in the beta-isomer content, which is the biologically active configuration required for therapeutic efficacy. This technical breakthrough not only simplifies the operational workflow but also aligns perfectly with the rigorous demands of modern Good Manufacturing Practice (GMP) environments, offering a reliable decitabine intermediate supplier pathway for global pharmaceutical developers seeking to optimize their API supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of decitabine has been plagued by significant stereochemical challenges that directly impact production costs and material throughput. Prior art, such as the routes reported by PISKALA or those utilizing acetyl-protected sugars, often relies on leaving groups that lack sufficient reactivity or stability, leading to prolonged reaction times and inconsistent yields. For instance, methods employing chlorine as a leaving group suffer from instability and storage difficulties, while alkoxy groups like methoxy exhibit low reactivity, necessitating harsher conditions that can degrade sensitive nucleobases. Furthermore, existing technologies, including those described in WO2009086687A1 and CN102070679A, typically result in a racemic mixture or a low proportion of the desired beta-isomer, with alpha-to-beta ratios hovering around 3:2 or even 1:1. This poor stereoselectivity forces manufacturers to rely on extensive and wasteful recrystallization processes or expensive chromatographic separations to isolate the active beta-form, creating a substantial bottleneck in cost reduction in API manufacturing and limiting the overall economic feasibility of large-scale production.
The Novel Approach
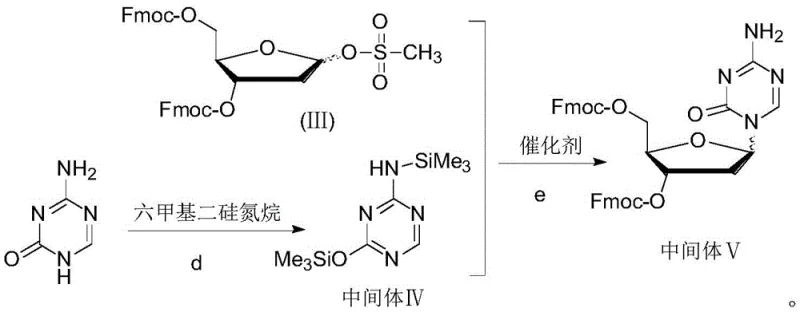
In stark contrast to these legacy methods, the novel approach disclosed in the patent utilizes a strategically designed 2-deoxy-D-ribose derivative featuring a mesylate leaving group and fluorenylmethoxycarbonyl (Fmoc) protecting groups. This specific structural configuration dramatically alters the electronic and steric environment of the glycosylation reaction, favoring the formation of the beta-anomer with unprecedented selectivity. The process involves coupling this highly reactive sugar derivative with 5-azacytosine that has been pre-activated by hexamethyldisilazane (HMDS), ensuring a smooth condensation under mild catalytic conditions. As illustrated in the reaction scheme below, this methodology shifts the isomeric ratio to an impressive alpha:beta of approximately 1:8, effectively solving the critical issue of low beta-isomer yield that has hindered previous attempts. By maximizing the formation of the target isomer at the source, this route minimizes downstream purification burdens and significantly enhances the overall material balance of the synthesis.
Mechanistic Insights into Stereoselective Glycosylation
The core of this technological advancement lies in the precise manipulation of the glycosidic bond formation through careful selection of protecting groups and leaving groups. The use of the mesylate group at the C-1 position of the ribose ring provides a superior leaving group capability compared to traditional acetyl or chloro substituents, facilitating a faster and more efficient nucleophilic attack by the silylated base. Simultaneously, the bulky Fmoc groups at the 3 and 5 positions exert a significant steric influence that directs the incoming nucleophile to attack from the beta-face, thereby suppressing the formation of the unwanted alpha-anomer. This synergistic effect between the high reactivity of the mesylate and the steric guidance of the Fmoc groups ensures that the reaction proceeds with high fidelity, producing a crude product that is already enriched with the desired stereochemistry. Such mechanistic control is essential for maintaining high-purity pharmaceutical intermediates, as it reduces the complexity of the impurity profile and simplifies the subsequent isolation steps.
Furthermore, the activation of 5-azacytosine via silylation with HMDS plays a crucial role in enhancing the nucleophilicity of the base while protecting its sensitive amino and carbonyl functionalities during the coupling process. The catalyst, preferably trimethylsilyl trifluoromethanesulfonate (TMSOTf), acts as a Lewis acid to promote the departure of the mesylate group and stabilize the oxocarbenium ion intermediate, further driving the reaction towards completion. This catalytic system operates effectively in polar aprotic solvents like acetonitrile at controlled low temperatures, typically between 0 to 10°C, which helps to minimize side reactions and degradation. The combination of these optimized reaction parameters results in a robust process that is not only chemically efficient but also highly reproducible, making it an ideal candidate for the commercial scale-up of complex nucleoside analogues where consistency is paramount.
How to Synthesize Decitabine Intermediate Efficiently
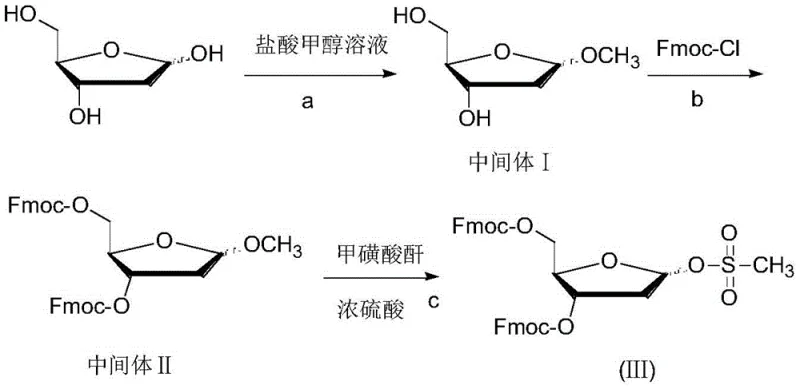
The synthesis protocol outlined in the patent provides a clear and actionable roadmap for producing the key intermediate with high purity and yield. The process begins with the preparation of the sugar donor, where 2-deoxy-D-ribose is first converted into a methyl glycoside and subsequently protected with Fmoc groups before undergoing mesylation to install the reactive leaving group. This precursor is then coupled with the HMDS-activated 5-azacytosine in the presence of a Lewis acid catalyst, followed by a straightforward workup procedure. The detailed standardized synthesis steps, including specific molar ratios, temperature controls, and solvent choices, are critical for replicating the high stereoselectivity reported in the examples. For R&D teams looking to implement this technology, adhering to the precise conditions described in the patent is essential to achieve the optimal alpha-to-beta ratio and ensure the quality of the final intermediate meets stringent regulatory standards.
- Synthesize the 2-deoxy-D-ribose derivative (III) by protecting 2-deoxy-D-ribose with Fmoc groups and introducing a mesylate leaving group at the C-1 position.
- Activate 5-azacytosine by refluxing with hexamethyldisilazane (HMDS) and ammonium sulfate to form the silylated intermediate (IV).
- Perform the coupling reaction between derivative (III) and activated base (IV) using TMSOTf as a catalyst in acetonitrile, followed by crystallization purification.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this patented method offers distinct advantages that translate directly into operational efficiency and risk mitigation. By fundamentally improving the stereoselectivity of the coupling reaction, the process eliminates the need for resource-intensive separation techniques that have traditionally driven up the cost of goods sold for decitabine. The ability to generate a crude product with a high proportion of the beta-isomer means that less raw material is wasted on unwanted byproducts, leading to substantial cost savings in raw material consumption and waste disposal. Additionally, the replacement of column chromatography with a simple crystallization step using common solvents like ethyl acetate and n-hexane drastically reduces the consumption of silica gel and organic solvents, further lowering the environmental footprint and operational expenses associated with the manufacturing process.
- Cost Reduction in Manufacturing: The elimination of silica gel column chromatography represents a major shift in processing economics, as chromatography is often a rate-limiting and expensive step in nucleoside synthesis. By relying on crystallization for purification, the process becomes significantly more amenable to continuous or large-batch processing, reducing labor hours and equipment downtime. The high yield of the desired beta-isomer also means that the effective throughput of the plant is increased without the need for additional capital investment, providing a clear path for cost reduction in pharmaceutical intermediate manufacturing through improved material efficiency and simplified downstream processing.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, such as 2-deoxy-D-ribose and 5-azacytosine, are commercially available and stable, ensuring a secure supply base for long-term production planning. The robustness of the reaction conditions, which tolerate mild temperatures and standard solvents, reduces the risk of batch failures due to sensitive operating parameters. This reliability is crucial for supply chain heads who need to guarantee consistent delivery schedules to downstream API manufacturers, as the process is less susceptible to the variability that often plagues more complex or hazardous synthetic routes involving unstable intermediates or extreme conditions.
- Scalability and Environmental Compliance: The use of benign solvents for crystallization and the avoidance of heavy metal catalysts or toxic reagents align well with modern environmental, health, and safety (EHS) standards. The simplicity of the workup procedure, involving basic aqueous washes and solvent evaporation, facilitates easy scale-up from pilot plant to multi-ton commercial production without requiring specialized containment or waste treatment infrastructure. This scalability ensures that the supply of high-purity decitabine intermediates can be rapidly expanded to meet market demand, supporting the growing clinical pipeline for decitabine-based therapies while maintaining compliance with increasingly strict global regulatory requirements for green chemistry practices.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and benefits of this synthesis method. These insights are derived directly from the experimental data and comparative examples provided in the patent documentation, offering clarity on how this approach differentiates itself from prior art. Understanding these nuances is vital for technical teams evaluating the feasibility of adopting this route for their own production needs, as it highlights the specific improvements in selectivity and purification that define the value proposition of this technology.
Q: How does this method improve the beta-isomer ratio compared to traditional routes?
A: Traditional methods using acetyl or methoxy leaving groups often yield alpha:beta ratios near 1:1 or 3:2. This patented method utilizes a mesylate leaving group combined with bulky Fmoc protecting groups, achieving a superior alpha:beta ratio of approximately 1:8, significantly reducing the burden of isomer separation.
Q: What purification method is used to avoid costly column chromatography?
A: The process employs a robust crystallization technique. After the coupling reaction, the crude product is dissolved in a benign ester solvent (like ethyl acetate) and precipitated into a poor solvent (like n-hexane), allowing for high-purity isolation without the need for silica gel columns.
Q: Which catalysts are suitable for the glycosylation step?
A: The patent identifies anhydrous stannic chloride (SnCl4) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) as effective catalysts, with TMSOTf being the preferred choice for optimizing reaction efficiency and selectivity in acetonitrile solvent.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Decitabine Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of life-saving oncology drugs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous volume requirements of global pharmaceutical partners. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs that utilize advanced analytical techniques to verify identity and impurity profiles. By leveraging advanced synthetic strategies like the one described in CN111377989B, we can offer a reliable decitabine intermediate supplier partnership that combines technical excellence with commercial reliability, ensuring your supply chain remains uninterrupted and cost-effective.
We invite you to engage with our technical procurement team to discuss how our capabilities can support your specific project needs. Whether you require a Customized Cost-Saving Analysis for your current supply chain or need to evaluate the feasibility of this novel synthetic route for your portfolio, we are ready to assist. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise in nucleoside chemistry can drive value and efficiency in your drug development programs.