Advanced Two-Step Synthesis of Azvudine: Enhancing Purity and Scalability for Global Pharmaceutical Supply Chains
Introduction to Next-Generation Azvudine Manufacturing
The global demand for effective antiretroviral therapies continues to drive innovation in the synthesis of complex nucleoside analogs, with Azvudine emerging as a critical component in HIV-1 treatment regimens. Patent CN114149475B discloses a transformative technological method for synthesizing Azvudine that fundamentally alters the traditional manufacturing landscape by replacing hazardous chlorination protocols with a safer, more efficient silyl ether aminolysis strategy. This intellectual property represents a significant leap forward for chemical manufacturers seeking to optimize their production lines for high-purity pharmaceutical intermediates while adhering to increasingly stringent environmental regulations. By leveraging a two-step sequence that begins with alkaline hydrolysis and concludes with a direct conversion of the pyrimidine 4-hydroxyl group, this process eliminates the need for toxic phosphorus oxychloride, thereby mitigating substantial safety risks and waste disposal costs associated with legacy synthetic routes. For industry stakeholders, understanding the nuances of this patented approach is essential for securing a competitive advantage in the supply of reliable antiviral drug substances.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Azvudine and related nucleoside analogs has been plagued by reliance on aggressive chlorinating agents, most notably phosphorus oxychloride, to activate the nucleobase for subsequent amination. These conventional pathways typically involve a multi-step sequence where the hydroxyl group is first converted to a chloride, followed by a separate aminolysis reaction to introduce the requisite amino functionality. This approach presents severe drawbacks, including the generation of large volumes of acidic wastewater containing phosphorus residues, which necessitates complex and expensive treatment protocols to meet environmental compliance standards. Furthermore, the handling of phosphorus oxychloride requires specialized corrosion-resistant equipment and rigorous safety measures due to its violent reactivity with moisture, creating significant operational bottlenecks and increasing the overall cost of goods sold. The complexity of these traditional routes often leads to lower overall yields due to cumulative losses across multiple isolation and purification stages, ultimately impacting the economic viability of large-scale production.
The Novel Approach
In stark contrast, the methodology outlined in the patent introduces a streamlined protocol that bypasses the chlorination step entirely, utilizing a direct silylation-aminolysis tandem reaction to achieve the desired structural transformation. By employing reagents such as hexamethyldisilazane (HMDS) in conjunction with simple amides like acetamide, the process activates the 4-position of the pyrimidine ring in situ, allowing for the direct displacement of the oxygen moiety with an amino group in a single operational step. This innovation not only reduces the total number of synthetic operations but also dramatically simplifies the workup procedure, as the byproducts are generally volatile silanols or easily removable salts rather than persistent phosphorus contaminants. The result is a cleaner reaction profile that facilitates easier purification, leading to higher purity final products with reduced impurity burdens, which is a critical quality attribute for regulatory approval in pharmaceutical manufacturing. This shift towards atom-economical and environmentally benign chemistry underscores a broader industry trend towards sustainable manufacturing practices without compromising on efficiency or product quality.
Mechanistic Insights into Silyl Ether Aminolysis
The core chemical innovation driving this process lies in the mechanistic pathway of the silyl ether aminolysis, which effectively converts a relatively inert hydroxyl group into a highly reactive leaving group capable of undergoing nucleophilic substitution by an amide nitrogen. In the presence of a silylating agent like HMDS, the 4-hydroxyl group of the uracil intermediate is rapidly transformed into a trimethylsilyl ether, which serves as an activated species susceptible to attack by the nitrogen lone pair of the acetamide. Under thermal conditions ranging from 100°C to 120°C, this interaction promotes the elimination of the silyl group and the formation of the carbon-nitrogen bond, effectively installing the exocyclic amine required for Azvudine's biological activity. This mechanism avoids the formation of unstable chloro-intermediates that are prone to hydrolysis or side reactions, thereby enhancing the robustness of the synthesis and ensuring consistent batch-to-batch reproducibility. The choice of amide solvent or reagent further influences the reaction kinetics, with acetamide providing an optimal balance of nucleophilicity and stability to drive the reaction to completion with minimal formation of over-alkylated or degraded byproducts.
Controlling the impurity profile in nucleoside synthesis is paramount, and this method offers distinct advantages in suppressing common degradation pathways associated with acid-sensitive glycosidic bonds. Traditional acidic chlorination conditions can sometimes lead to depurination or cleavage of the sugar-base linkage, particularly in fluorinated nucleosides where the electronic properties of the sugar ring are altered. By maintaining neutral to slightly basic conditions during the silylation and aminolysis phases, the integrity of the glycosidic bond is preserved, minimizing the formation of free base impurities that are difficult to separate from the final product. Additionally, the initial alkaline hydrolysis step is carefully controlled to selectively remove the ester protecting group on the sugar ring without affecting the azido functionality or the fluorine substituent, demonstrating a high degree of chemoselectivity. This precise control over reaction parameters ensures that the final Azvudine product meets stringent purity specifications, reducing the need for extensive chromatographic purification and lowering the overall processing time and solvent consumption.
How to Synthesize Azvudine Efficiently
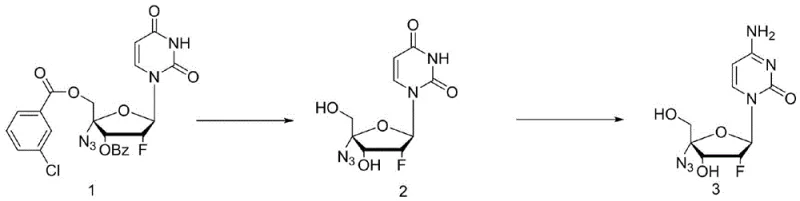
The practical implementation of this synthesis route involves a straightforward two-stage protocol that is amenable to standard reactor configurations found in most fine chemical facilities. The process begins with the deprotection of the starting material, a 2'-fluoro-4'-azido-uracil nucleoside derivative bearing a benzoate or similar ester group on the sugar moiety. This step is executed by treating the substrate with a mild base such as ammonia in methanol or sodium methoxide in ethanol at moderate temperatures, typically between 50°C and 60°C, to cleave the ester bond and reveal the free hydroxyl groups necessary for the subsequent transformation. Following isolation of the deprotected intermediate, the material is subjected to the key silylation-aminolysis reaction by heating with excess hexamethyldisilazane and acetamide, driving the conversion to the final amino-nucleoside. The detailed standardized synthesis steps, including specific molar ratios, quenching procedures, and crystallization parameters, are provided in the guide below to ensure reproducible results.
- Perform alkaline hydrolysis on the protected 2'-fluoro-4'-azido-uracil nucleoside using ammonia or alkoxides in alcohol solvents at 50-60°C to remove the sugar ring protecting group.
- React the resulting intermediate with a silyl ether reagent like hexamethyldisilazane and an amide such as acetamide at 100-120°C to directly convert the 4-hydroxyl group to an amino group.
- Quench the reaction with methanol, filter the crude solid, and purify via recrystallization in ethanol-water mixtures to obtain high-purity Azvudine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers compelling strategic benefits that extend beyond mere technical feasibility, directly impacting the bottom line through operational efficiencies and risk mitigation. The elimination of phosphorus oxychloride from the supply chain removes a significant logistical burden, as this reagent is subject to strict transportation regulations and requires specialized storage infrastructure, the removal of which simplifies facility management and reduces insurance premiums. Furthermore, the reduction in reaction steps translates to shorter cycle times and lower labor costs per kilogram of produced API intermediate, allowing manufacturers to respond more agilely to market demand fluctuations. The use of commodity chemicals like acetamide and HMDS, which are widely available from multiple global suppliers, ensures a resilient supply chain that is less vulnerable to the geopolitical or production disruptions that often affect specialty reagents. This robustness in raw material sourcing is a critical factor for maintaining continuous production schedules and meeting delivery commitments to downstream pharmaceutical partners.
- Cost Reduction in Manufacturing: The economic argument for switching to this process is anchored in the substantial simplification of the workflow, which inherently lowers variable costs associated with reagents, energy, and waste treatment. By consolidating the activation and amination into a single thermal step, the process reduces the consumption of solvents and the energy required for heating and cooling cycles across multiple stages. The avoidance of phosphorus-based waste streams eliminates the need for costly neutralization and disposal services, which can represent a significant portion of operating expenses in traditional nucleoside manufacturing. Additionally, the improved yield stability observed during scale-up means that less starting material is wasted, maximizing the return on investment for every batch produced and contributing to a more favorable cost structure for the final commercial product.
- Enhanced Supply Chain Reliability: From a logistics perspective, the reliance on stable, non-hazardous reagents enhances the predictability of the supply chain, reducing the likelihood of delays caused by regulatory hold-ups or safety incidents. The simplified process flow also decreases the dependency on complex, custom-manufactured intermediates, allowing for greater flexibility in sourcing and inventory management. This reliability is crucial for long-term supply agreements with major pharmaceutical companies, where consistency and on-time delivery are non-negotiable requirements. By adopting a method that utilizes common industrial solvents and reagents, manufacturers can leverage existing supplier relationships and bulk purchasing power to further optimize procurement costs and secure favorable terms.
- Scalability and Environmental Compliance: The environmental footprint of this synthesis is markedly lower than that of conventional methods, aligning with the growing corporate mandate for sustainable manufacturing practices and green chemistry principles. The reduction in hazardous waste generation simplifies the permitting process for facility expansion and minimizes the risk of environmental liabilities, making it an attractive option for companies looking to future-proof their operations. The process demonstrates excellent scalability, with pilot data indicating that yield and purity remain consistent even when transitioning from laboratory to multi-ton production scales. This scalability ensures that the technology can support the growing global demand for Azvudine without requiring disproportionate increases in capital expenditure or environmental remediation efforts.
Frequently Asked Questions (FAQ)
To address common inquiries regarding the technical and commercial implications of this synthesis method, we have compiled a set of answers based on the specific details disclosed in the patent literature. These responses clarify the operational parameters and highlight the distinct advantages of this route over legacy technologies, providing decision-makers with the clarity needed to evaluate its potential integration into their manufacturing portfolios. Understanding these nuances is vital for assessing the feasibility of technology transfer and the anticipated return on investment for process optimization initiatives.
Q: How does this new Azvudine synthesis method improve upon traditional chlorination routes?
A: Traditional methods rely on phosphorus oxychloride for chlorination followed by aminolysis, which generates hazardous waste and requires strict safety controls. This novel patent methodology bypasses the chlorination step entirely by utilizing hexamethyldisilazane and amides for direct conversion, significantly reducing environmental impact and operational risk while simplifying the purification workflow.
Q: What are the key reagents required for the silyl ether aminolysis step?
A: The critical second step involves heating the deprotected intermediate with a silylating agent such as hexamethyldisilazane (HMDS) or N,O-bis(trimethylsilyl)trifluoroacetamide in the presence of an amide source like acetamide or formamide. This combination facilitates the activation of the 4-hydroxyl position and its subsequent displacement by the amino group under thermal conditions.
Q: Is this process suitable for large-scale commercial production of antiviral intermediates?
A: Yes, the process is specifically designed for scalability. By eliminating the use of corrosive phosphorus oxychloride and reducing the total number of reaction steps, the method offers stable yields during amplification. The use of common organic solvents and reagents ensures that supply chain continuity is maintained, making it highly viable for industrial manufacturing of high-purity pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Azvudine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic methodologies to maintain competitiveness in the fast-evolving landscape of antiviral drug manufacturing. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative patents like CN114149475B can be seamlessly translated into robust, GMP-compliant manufacturing processes. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify every batch. Our dedication to quality and safety makes us an ideal partner for pharmaceutical companies seeking to secure a stable and compliant supply of complex nucleoside analogs.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis route can be tailored to your specific production needs and volume requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the potential economic benefits of switching to this greener, more efficient process. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that will enhance your supply chain resilience and drive long-term value for your organization.