Scaling Bivalirudin Production: A Technical Breakthrough in Liquid Phase Peptide Synthesis
The pharmaceutical industry continuously seeks robust manufacturing routes for complex polypeptides, and the liquid phase synthesis method for Bivalirudin disclosed in patent CN101475631B represents a significant paradigm shift from traditional solid-phase techniques. This technology addresses the critical bottlenecks of scalability and safety by employing a strategic fragment condensation approach, dividing the 20-amino acid sequence into three manageable, fully protected segments. Unlike conventional methods that rely on costly resin supports and hazardous cleavage reagents, this novel pathway utilizes standard solution-phase chemistry to achieve high-purity intermediates through rigorous purification at every stage. The process culminates in the assembly of the full peptide chain followed by a mild global deprotection step, ensuring that the final active pharmaceutical ingredient meets stringent quality specifications without the environmental burden of toxic waste streams. For procurement and technical teams, this methodology offers a compelling value proposition by decoupling production capacity from the limitations of specialized peptide synthesizers.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional solid-phase peptide synthesis (SPPS) for large molecules like Bivalirudin faces inherent challenges that severely impact commercial viability and operational safety. The reliance on specialized resins, such as CTC or Wang resin, introduces a substantial variable cost that scales linearly with production volume, making mass manufacturing economically inefficient compared to small-batch laboratory synthesis. Furthermore, the final cleavage step in many established protocols necessitates the use of anhydrous hydrogen fluoride (HF), a substance of severe toxicity and corrosiveness that requires dedicated, expensive containment infrastructure and poses significant risks to personnel and the environment. Additionally, SPPS often suffers from the accumulation of deletion sequences and incomplete couplings as the chain lengthens, leading to complex impurity profiles that are difficult to resolve without extensive and yield-reducing purification efforts. These factors collectively create a high barrier to entry for reliable pharmaceutical intermediates supplier operations aiming to produce cardiovascular therapeutics at an industrial scale.
The Novel Approach
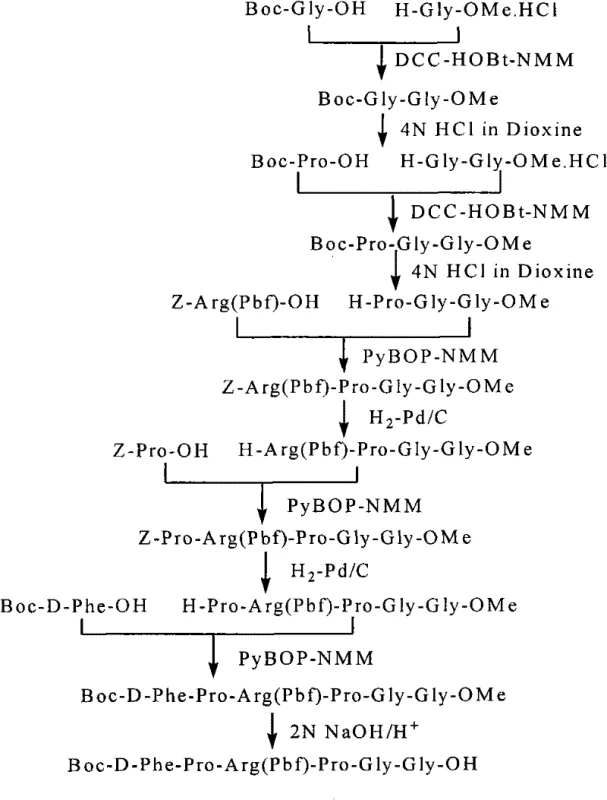
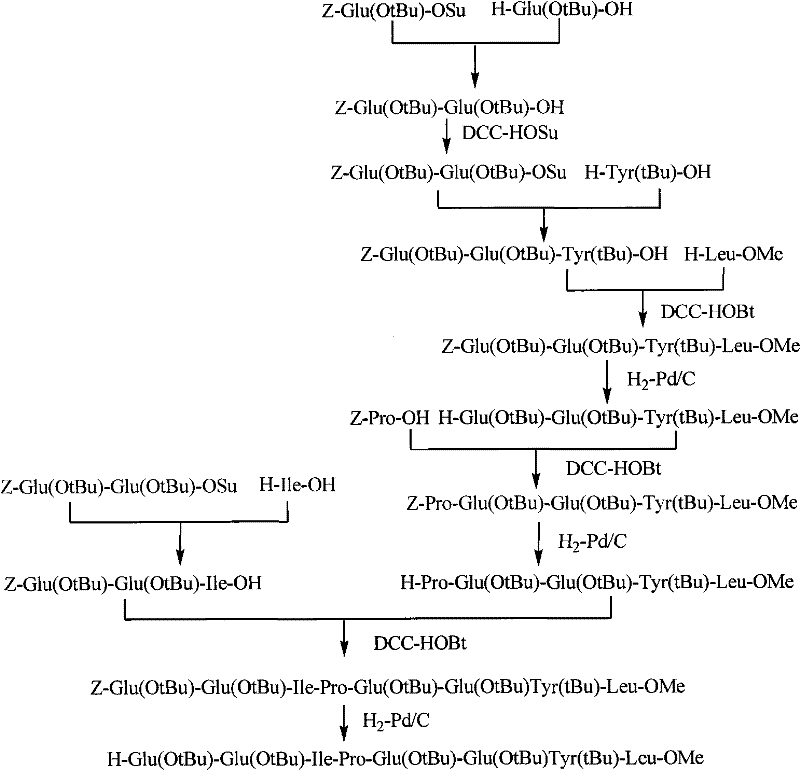
The liquid phase synthesis route described in the patent data overcomes these hurdles by adopting a convergent fragment condensation strategy that prioritizes modularity and purification control. By synthesizing three distinct fragments—an N-terminal 6-peptide, a middle 6-peptide, and a C-terminal 8-peptide—independently, the process allows for the isolation and purification of high-quality building blocks before the final assembly. This method eliminates the need for solid supports entirely, thereby removing the associated resin costs and the physical limitations of swelling and diffusion within a resin matrix. The use of standard coupling reagents like PyBOP, DCC, and HOBt in solution phase facilitates better mixing and reaction kinetics, while the avoidance of HF cleavage in favor of TFA-TIS deprotection drastically simplifies the safety protocols and waste management requirements. This approach not only enhances the feasibility of cost reduction in API manufacturing but also ensures a more consistent supply chain by utilizing widely available chemical inputs and standard reactor equipment.
Mechanistic Insights into Fragment Condensation and Orthogonal Protection
The success of this liquid phase synthesis hinges on a meticulously designed orthogonal protection strategy that safeguards reactive side chains while enabling selective peptide bond formation. For instance, the Arginine residue is protected with the Pbf (2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl) group, which is stable under basic coupling conditions but readily cleaved by acid, preventing guanidine side reactions. Similarly, acidic residues like Aspartic acid and Glutamic acid are masked as tert-butyl esters (OtBu), and Asparagine is protected with a Trityl (Trt) group to prevent dehydration and cyclization side reactions. The coupling reactions utilize activated ester mechanisms, where reagents like PyBOP or DCC activate the carboxyl group of the incoming amino acid or fragment, forming an reactive intermediate that is attacked by the free amine of the growing chain. The inclusion of additives like HOBt or HOSu is critical in suppressing racemization, ensuring that the stereochemical integrity of the chiral centers is maintained throughout the multi-step synthesis, which is paramount for the biological activity of the final thrombin inhibitor.
Impurity control is rigorously managed through the intermediate purification of each fragment, a distinct advantage over linear solid-phase synthesis where impurities propagate. After each coupling step, such as the formation of the Z-Glu(OtBu)-Glu(OtBu)-Tyr(tBu)-Leu-OMe intermediate, the product is subjected to workup procedures involving acid/base washes and silica gel column chromatography. This allows for the removal of unreacted starting materials, urea byproducts from carbodiimide reagents, and any deletion peptides before they can complicate the subsequent condensation. The final global deprotection using a TFA-TIS mixture efficiently removes all acid-labile protecting groups (Boc, Pbf, OtBu, Trt) in a single operation, yielding the crude peptide which is then polished via high-performance liquid chromatography (HPLC). This multi-stage purification protocol ensures that the final product achieves the high purity levels required for clinical application, minimizing the risk of immunogenic responses caused by peptide aggregates or truncated sequences.
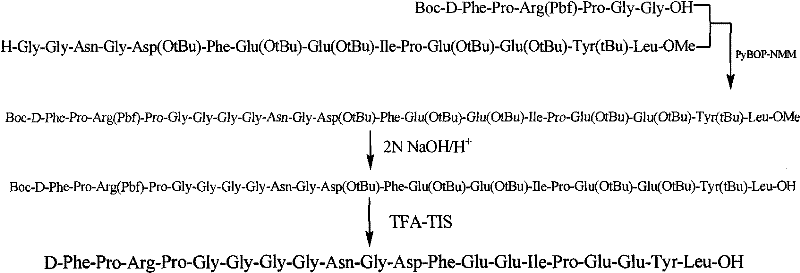
How to Synthesize Bivalirudin Efficiently
The synthesis of Bivalirudin via this liquid phase method involves a logical progression of fragment assembly that balances reaction efficiency with ease of purification. The process begins with the independent preparation of the three key segments using standard peptide coupling chemistry in organic solvents like THF or DMF. Once the individual fragments are secured and purified, they are sequentially condensed, first joining the middle and C-terminal segments to form a larger 14-peptide unit, and finally coupling this with the N-terminal segment to complete the 20-amino acid backbone. Detailed standardized synthetic steps see the guide below.
- Synthesize three protected fragments separately: N-terminal 6-peptide (Segment A), middle 6-peptide (Fragment B), and C-terminal 8-peptide (Fragment C) using standard coupling reagents like PyBOP and DCC.
- Condense Fragment B and Fragment C to form the 14-peptide intermediate (Fragment D), followed by purification via silica gel column chromatography.
- Couple Segment A with Fragment D to obtain the fully protected 20-peptide, then perform global deprotection using TFA-TIS and purify via HPLC to yield pure Bivalirudin.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this liquid phase synthesis route offers transformative benefits that directly impact the bottom line and operational resilience. The elimination of expensive solid-phase resins and the reduction in the excess usage of protected amino acids significantly lower the raw material costs per kilogram of finished product. Furthermore, the ability to recycle common organic solvents used in the liquid phase reactions, as opposed to the large volumes of washing solvents required for resin swelling and cleaning, contributes to substantial cost savings and aligns with green chemistry initiatives. The process is inherently safer due to the absence of hydrogen fluoride, reducing the need for specialized containment facilities and lowering insurance and compliance overheads. This makes the technology particularly attractive for establishing reliable supply chains in regions with strict environmental regulations.
- Cost Reduction in Manufacturing: The economic advantages of this method are driven by the removal of resin costs and the optimization of reagent stoichiometry. In solid-phase synthesis, a large excess of amino acids is often required to drive reactions to completion on a solid support, leading to significant waste; conversely, liquid phase coupling allows for more precise control over molar ratios. Additionally, the avoidance of toxic HF cleavage eliminates the need for specialized tetrafluoroethylene equipment and the associated high maintenance and disposal costs. These factors combine to drastically simplify the cost structure, making the commercial scale-up of complex pharmaceutical intermediates much more financially viable.
- Enhanced Supply Chain Reliability: The reliance on standard chemical reagents and solvents rather than proprietary resins enhances supply security. Resins can sometimes face availability bottlenecks or batch-to-batch variability that impacts yield, whereas the chemicals used in this liquid phase route (such as DCC, PyBOP, and standard protected amino acids) are commodity items available from multiple global suppliers. This diversification of the supply base reduces the risk of production stoppages due to single-source dependency. Moreover, the simplified equipment requirements mean that production can be easily transferred between different manufacturing sites without the need for custom-built peptide synthesizers, ensuring continuity of supply even during geopolitical or logistical disruptions.
- Scalability and Environmental Compliance: Scaling liquid phase reactions is a well-understood engineering challenge that can be addressed with standard stainless steel reactors, unlike solid-phase processes which often face mixing and filtration issues at large scales. The process generates less hazardous waste, particularly by avoiding fluorine-containing effluents from HF cleavage, which simplifies wastewater treatment and reduces the environmental footprint. This ease of scale-up from pilot batches to multi-ton production ensures that the manufacturing capacity can grow in tandem with market demand for Bivalirudin, supporting long-term commercial agreements without the need for prohibitive capital expenditure on specialized infrastructure.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the liquid phase production of Bivalirudin, based on the specific methodologies and advantages outlined in the patent literature. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this synthesis route for their own manufacturing pipelines or for procurement specialists assessing the quality and stability of the supply source. The answers provided reflect the consensus on best practices for peptide synthesis and the specific innovations introduced by this fragment condensation strategy.
Q: Why is liquid phase synthesis preferred over solid phase for Bivalirudin mass production?
A: Liquid phase synthesis eliminates the need for expensive specialty resins and excessive amounts of protected amino acids required in solid phase synthesis. It avoids the use of highly toxic hydrogen fluoride (HF) for cleavage, significantly improving safety and reducing environmental compliance costs while facilitating easier scale-up in standard reactors.
Q: How does the 3-fragment condensation strategy improve purity?
A: By synthesizing smaller fragments (6-peptide, 6-peptide, and 8-peptide) individually, impurities can be removed at each stage using crystallization or column chromatography before the final assembly. This prevents the accumulation of deletion sequences and side products that often plague long linear solid-phase syntheses, resulting in a higher quality crude product prior to HPLC purification.
Q: What protecting groups are used to prevent side reactions during synthesis?
A: The process utilizes an orthogonal protection strategy including Boc for the N-terminus, Pbf for Arginine, OtBu for Aspartic and Glutamic acids, and Trt for Asparagine. This ensures that side chains remain protected during coupling steps and can be globally removed in the final stage using TFA without affecting the peptide backbone.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Bivalirudin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthesis technologies to meet the growing global demand for cardiovascular therapeutics. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless and efficient. We are committed to maintaining stringent purity specifications through our rigorous QC labs, utilizing state-of-the-art analytical methods to verify the identity and quality of every batch of Bivalirudin intermediate and API we produce. Our facility is equipped to handle the specific requirements of liquid phase peptide synthesis, providing a secure and compliant environment for the manufacture of high-value pharmaceutical ingredients.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific supply chain needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to our liquid-phase manufactured intermediates. We encourage you to contact us directly to obtain specific COA data and route feasibility assessments tailored to your project timelines, ensuring that your development programs proceed with the highest quality materials available in the market today.