Advanced Bivalirudin Manufacturing: High-Purity Peptide Synthesis for Global Pharma
The pharmaceutical industry continuously demands higher standards for polypeptide therapeutics, particularly for anticoagulants like Bivalirudin where impurity profiles directly impact patient safety. Patent CN102286076A introduces a transformative preparation method that addresses the longstanding challenges associated with solid-phase peptide synthesis (SPPS) of complex sequences. This technical breakthrough focuses on the strategic insertion of a tetraglycine fragment, specifically Fmoc-Gly-Gly-Gly-Gly-OH, to bypass the kinetic limitations of stepwise amino acid coupling. By shifting from individual glycine additions to a fragment condensation approach, the method fundamentally alters the impurity landscape, offering a pathway to achieve purity levels exceeding 99.5%. For R&D directors and procurement specialists, this represents a critical evolution in manufacturing reliability, ensuring that the final active pharmaceutical ingredient meets the most stringent regulatory specifications while optimizing the overall production workflow for commercial viability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional solid-phase synthesis of Bivalirudin typically involves the sequential addition of individual glycine residues to build the tetraglycine bridge connecting the N-terminal and C-terminal domains. This stepwise approach is inherently prone to incomplete coupling reactions and racemization, which generate a complex spectrum of deletion and insertion impurities. Specifically, the repetitive nature of glycine coupling often leads to the formation of [+1Gly] and [+2Gly] variants, as well as deletion sequences like [1Gly] and [2Gly]. These impurities possess physicochemical properties, such as polarity and hydrophobicity, that are remarkably similar to the target molecule, making their removal via standard chromatographic techniques extremely difficult and costly. Consequently, conventional methods often struggle to consistently achieve the high purity required for clinical applications, resulting in significant yield loss during the purification stage and increased manufacturing costs due to the need for extensive downstream processing.
The Novel Approach
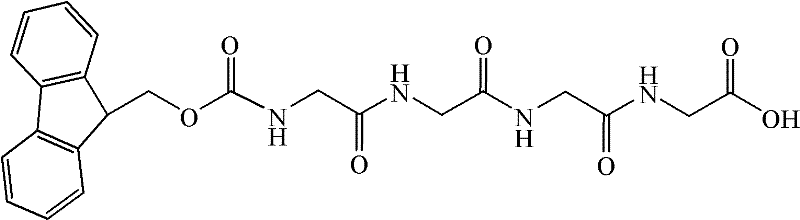
The innovative strategy outlined in the patent data circumvents these kinetic bottlenecks by utilizing a pre-synthesized, fully protected tetraglycine fragment, Fmoc-Gly-Gly-Gly-Gly-OH, as a single building block. This fragment condensation technique drastically reduces the number of coupling cycles required for this specific region of the peptide chain, thereby minimizing the cumulative risk of side reactions. By introducing the entire four-glycine sequence in one highly efficient coupling step, the formation of intermediate deletion sequences is effectively eliminated at the source. This approach not only streamlines the synthesis timeline but also significantly simplifies the impurity profile of the crude peptide, allowing for more efficient purification. For supply chain managers, this translates to a more robust process with higher predictability, reducing the risk of batch failures and ensuring a consistent supply of high-quality material for downstream formulation.
Mechanistic Insights into Fmoc-SPPS Fragment Condensation
The core of this synthesis lies in the precise management of steric hindrance and coupling efficiency during the solid-phase assembly. In standard Fmoc chemistry, the repetitive addition of glycine can lead to aggregation on the resin, which shields reactive amino groups and results in incomplete acylation. The use of the tetraglycine fragment mitigates this by presenting a pre-organized structure that couples more readily with the growing peptide chain on the resin. The reaction utilizes potent coupling reagents such as DIC (N,N'-Diisopropylcarbodiimide) and activators like HOBt (1-Hydroxybenzotriazole) to ensure rapid and complete activation of the carboxyl group. This mechanistic advantage ensures that the critical tetraglycine bridge is formed with high fidelity, preserving the structural integrity required for the molecule's biological activity as a direct thrombin inhibitor.
Furthermore, the control of side-chain protecting groups plays a pivotal role in the final purity of the Bivalirudin product. The patent specifies the use of acid-labile protecting groups such as Pbf for arginine and OtBu for aspartic and glutamic acids, which are stable during the base-mediated Fmoc deprotection cycles but are efficiently removed during the final acidolysis step. This orthogonality is crucial for preventing premature deprotection or side reactions that could lead to branched peptides or truncated sequences. The final cleavage cocktail, comprising trifluoroacetic acid (TFA), ethanedithiol (EDT), and water, is optimized to simultaneously cleave the peptide from the resin and remove all side-chain protecting groups without inducing unwanted modifications. This precise control over the deprotection mechanism ensures that the final crude product is chemically homogeneous, facilitating the subsequent high-performance liquid chromatography (HPLC) purification to achieve the target specification of less than 0.2% for any single impurity.
How to Synthesize Bivalirudin Efficiently
The synthesis protocol begins with the preparation of the initial resin, typically Fmoc-Leu-Wang or Fmoc-Leu-2-Cl-Trt resin, which serves as the anchor for the C-terminal amino acid. Following the initial loading, the peptide chain is elongated using standard Fmoc-SPPS cycles until the position preceding the tetraglycine sequence is reached. At this critical juncture, the pre-synthesized Fmoc-Gly-Gly-Gly-Gly-OH fragment is introduced, coupled under optimized conditions to ensure quantitative reaction. The synthesis continues with the addition of the remaining N-terminal amino acids, followed by a final global deprotection and cleavage step. The detailed standardized synthesis steps see the guide below.
- Prepare Fmoc-Leu-Wang resin and sequentially couple protected amino acids using DIC/HOBt activation, ensuring strict stoichiometric control.
- Insert the critical Fmoc-Gly-Gly-Gly-Gly-OH fragment in a single coupling step to prevent deletion sequences and Gly-insertion impurities.
- Perform acidolysis using a TFA/EDT/Water cocktail followed by preparative HPLC purification to achieve >99.5% purity.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this fragment-based synthesis route offers substantial advantages in terms of cost structure and supply chain resilience. By significantly reducing the complexity of the impurity profile, the manufacturing process requires less aggressive and fewer purification cycles to meet pharmacopeial standards. This reduction in downstream processing directly correlates to lower solvent consumption, reduced waste generation, and shorter production cycles, all of which contribute to a more sustainable and cost-effective manufacturing operation. For procurement managers, this efficiency gain means a more stable pricing model and reduced exposure to the volatility of raw material costs associated with excessive solvent and resin usage.
- Cost Reduction in Manufacturing: The elimination of stepwise glycine coupling reduces the consumption of expensive coupling reagents and protected amino acids, as fewer activation cycles are required to build the peptide chain. Furthermore, the improved crude purity minimizes the loss of product during the purification phase, leading to a higher overall yield of the final active ingredient. This process intensification allows for significant cost optimization in API manufacturing without compromising on quality, providing a competitive edge in the global market for anticoagulant therapeutics.
- Enhanced Supply Chain Reliability: The robustness of the fragment condensation method reduces the likelihood of batch-to-batch variability, which is a common risk in complex peptide synthesis. A more predictable synthesis outcome ensures that production schedules can be met with greater certainty, reducing lead time for high-purity polypeptides. This reliability is critical for pharmaceutical partners who require a consistent supply of intermediates to maintain their own formulation and packaging timelines, thereby strengthening the overall resilience of the pharmaceutical supply chain.
- Scalability and Environmental Compliance: The simplified process flow is inherently more scalable, allowing for the commercial scale-up of complex polypeptides from laboratory to multi-ton production with minimal process re-engineering. Additionally, the reduction in solvent usage and waste generation aligns with increasingly stringent environmental regulations, reducing the burden of waste disposal and compliance monitoring. This environmental efficiency not only lowers operational costs but also enhances the corporate sustainability profile of the manufacturing partner.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of Bivalirudin using this advanced methodology. These insights are derived directly from the patent specifications and are intended to provide clarity on the process capabilities and quality assurances. Understanding these details is essential for technical teams evaluating the feasibility of this route for their specific supply chain requirements.
Q: How does the tetraglycine fragment improve Bivalirudin purity?
A: By using a pre-synthesized Fmoc-Gly-Gly-Gly-Gly-OH fragment, the method eliminates the risk of stepwise coupling errors that typically generate [+1Gly] and [+2Gly] impurities, which are difficult to separate due to similar polarity.
Q: What are the critical impurities controlled in this process?
A: The process specifically targets the reduction of deletion sequences and Gly-insertion variants, ensuring single impurity levels remain below 0.2% and total purity exceeds 99.5%.
Q: Is this synthesis method scalable for commercial production?
A: Yes, the simplified coupling steps and robust purification protocol are designed for commercial scale-up, offering significant advantages in process efficiency and batch-to-batch consistency.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Bivalirudin Supplier
NINGBO INNO PHARMCHEM stands at the forefront of peptide manufacturing, leveraging advanced synthesis technologies like the fragment condensation method to deliver superior quality Bivalirudin. Our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production ensures that we can meet the rigorous demands of global pharmaceutical clients. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the highest international standards, providing our partners with the confidence needed to advance their clinical and commercial programs.
We invite you to collaborate with us to optimize your supply chain for this critical anticoagulant. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our technical expertise can drive value and efficiency in your Bivalirudin sourcing strategy.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →