Advanced Solid-Phase Synthesis of Bivalirudin for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust manufacturing pathways for complex anticoagulant peptides, and patent CN101906150B presents a significant breakthrough in the preparation of Bivalirudin. This document details a sophisticated hybrid synthesis strategy that merges the precision of liquid-phase peptide synthesis with the efficiency of solid-phase techniques. Traditional methods often struggle with the accumulation of deletion sequences and difficult couplings, particularly in glycine-rich regions, leading to costly purification burdens. The disclosed invention addresses these critical bottlenecks by introducing pre-formed peptide fragments, specifically Fmoc-Asn(Trt)-Gly-OH and Fmoc-Gly-Gly-Gly-Gly-OH, into the solid-phase workflow. This approach not only streamlines the synthetic route but also demonstrably enhances the final product's purity profile by suppressing the formation of [Bivalirudin-Gly] and [Bivalirudin+Gly] impurities. For R&D directors and procurement specialists, understanding this mechanistic shift is vital for securing a reliable Bivalirudin supplier capable of delivering high-quality API intermediates at a competitive cost structure.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Conventional solid-phase peptide synthesis (SPPS) for long-chain peptides like Bivalirudin often encounters significant hurdles related to coupling efficiency and steric hindrance as the chain elongates. When amino acids are added sequentially one by one on the resin, the probability of incomplete reactions increases, leading to a complex mixture of deletion peptides that are structurally similar to the target molecule. Specifically, the poly-glycine sequences found in Bivalirudin are prone to aggregation and difficult coupling, which traditional linear SPPS fails to address effectively. Furthermore, prior art methods, such as those disclosed in US20070093423A, rely on fragment condensation in solution that can be operationally complex and yield limited product purity. The accumulation of impurities necessitates extensive downstream purification, often involving preparative HPLC, which drastically reduces overall yield and escalates manufacturing costs. Additionally, the use of hazardous solvents like diethyl ether for precipitation in older protocols poses significant safety risks in a commercial plant environment, complicating regulatory compliance and operational safety standards.
The Novel Approach
The novel approach outlined in patent CN101906150B fundamentally restructures the synthesis by integrating liquid-phase synthesized fragments directly onto the solid support. Instead of adding single glycine residues repeatedly, the method employs a tetra-glycine fragment (Fmoc-Gly-Gly-Gly-Gly-OH) and an Asn-Gly dipeptide fragment (Fmoc-Asn(Trt)-Gly-OH) that are rigorously purified in the liquid phase before being coupled to the resin-bound peptide chain. This strategy effectively bypasses the most error-prone steps of linear synthesis, ensuring that the difficult sequences are already formed with high fidelity before attachment. By condensing these high-purity fragments onto the polypeptide resin shown in Formula V and Formula VI, the process minimizes the generation of deletion sequences right at the source. This results in a crude peptide with significantly higher purity, reportedly reaching 80%-91%, which simplifies the final purification steps. Moreover, the substitution of diethyl ether with methyl tert-butyl ether (MTBE) for precipitation enhances the safety profile, making the process more amenable to large-scale commercial operations without compromising on yield or quality.
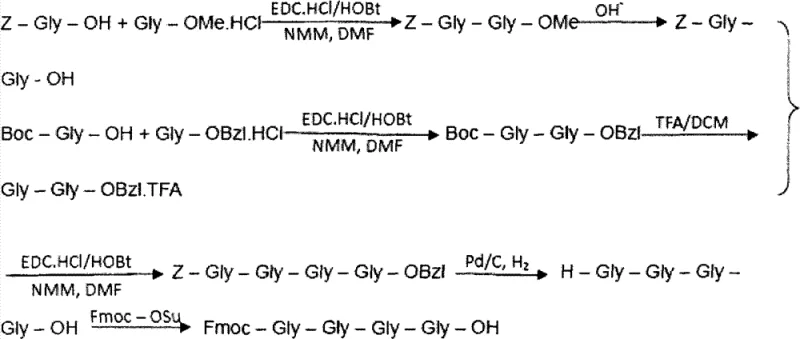
Mechanistic Insights into Hybrid Fragment Condensation
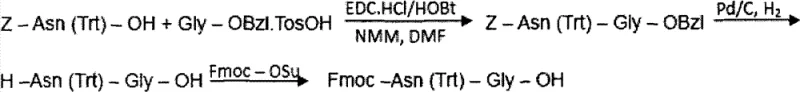
The core mechanistic advantage of this synthesis lies in the strategic deployment of orthogonal protecting groups and the precise control of condensation reactions. The liquid-phase synthesis of the Fmoc-Gly-Gly-Gly-Gly-OH fragment involves the condensation of Z-Gly-Gly-OH with H-Gly-Gly-OBzl, followed by hydrogenolysis and Fmoc protection. This multi-step liquid phase preparation ensures that the glycine cluster, which is notoriously difficult to couple on solid phase due to potential aggregation, is introduced as a single, high-integrity unit. Similarly, the Fmoc-Asn(Trt)-Gly-OH fragment is prepared via liquid-phase condensation of Z-Asn(Trt)-OH and H-Gly-OBzl, followed by deprotection and Fmoc introduction. The use of the Trt (trityl) protecting group on the Asn side chain is crucial as it prevents side reactions during the subsequent solid-phase elongation. When these fragments are coupled to the resin, the reaction is monitored using the ninhydrin (Kaiser) test to ensure completeness, with Chloranil testing employed specifically after proline couplings to detect secondary amines accurately.
Impurity control is further enhanced by the specific composition of the deprotection reagent used throughout the solid-phase cycle. The patent specifies a deprotection mixture containing piperidine and DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), which acts synergistically to remove the Fmoc group rapidly and cleanly. The inclusion of DBU helps to prevent the formation of aspartimide byproducts, a common issue in aspartic acid-containing peptides, while additives like HOBt or HOOBT suppress racemization during activation. The final cleavage step utilizes a cocktail of trifluoroacetic acid (TFA), triisopropylsilane (TIS), and water in a ratio of 95:2.5:2.5. This specific scavenger system effectively captures carbocations released during the removal of acid-labile protecting groups (such as tBu and Pbf), preventing them from alkylating the peptide backbone. This rigorous control over reaction conditions and reagent stoichiometry is what allows the process to achieve impurity levels of [Bivalirudin-Gly] below 0.6% and [Bivalirudin+Gly] below 0.2%, a specification that is critical for regulatory approval.
How to Synthesize Bivalirudin Efficiently
The synthesis of Bivalirudin via this hybrid method requires precise execution of both liquid-phase fragment preparation and solid-phase elongation. The process begins with the independent synthesis of the critical glycine and asparagine fragments in solution, where reaction conditions can be tightly optimized for maximum yield and purity. Once these fragments are secured, they are introduced into the solid-phase reactor containing the growing peptide chain attached to Wang resin. The coupling of these larger fragments requires careful monitoring to ensure full conversion, utilizing reagents like DIC and HOBt to activate the carboxyl termini. The detailed standardized synthesis steps, including specific molar equivalents, reaction times, and washing protocols required to replicate this high-efficiency route, are outlined in the guide below.
- Synthesize key peptide fragments (Fmoc-Gly-Gly-Gly-Gly-OH and Fmoc-Asn(Trt)-Gly-OH) using liquid-phase condensation methods to ensure high purity before resin attachment.
- Load the C-terminal amino acid (Leucine) onto Wang resin and perform sequential solid-phase coupling from the C-terminus to the N-terminus up to the Asp11 position.
- Couple the pre-synthesized liquid-phase fragments onto the growing peptide chain on the resin, followed by final N-terminal coupling and cleavage using TFA/TIS/Water.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthesis method offers tangible benefits that extend beyond mere technical elegance. The primary advantage is the substantial reduction in manufacturing costs driven by the simplification of the purification process. By minimizing the formation of closely related impurities early in the synthesis, the burden on downstream chromatography is significantly lessened, leading to higher overall recovery rates of the final API. The patent documentation explicitly claims that comparing this technology with traditional methods can reduce costs by approximately 50%, a figure driven by reduced solvent consumption, fewer processing steps, and higher yields. This cost efficiency translates directly into a more competitive pricing structure for the final Bivalirudin product, allowing pharmaceutical companies to optimize their COGS (Cost of Goods Sold) without sacrificing quality standards.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the reduction in solvent usage for purification contribute to a leaner manufacturing budget. The ability to use MTBE instead of highly volatile and hazardous diethyl ether also reduces safety infrastructure costs and insurance premiums associated with handling flammable solvents. Furthermore, the higher crude purity means less resin and reagent waste per kilogram of final product, optimizing the utilization of raw materials. This economic efficiency makes the process highly attractive for generic drug manufacturers looking to enter the antithrombotic market with a cost-effective alternative to branded formulations.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route ensures a more stable supply of Bivalirudin intermediates. Because the critical fragments are synthesized in the liquid phase where quality control is easier to enforce, the risk of batch failure during the expensive solid-phase stage is minimized. This reliability is crucial for maintaining continuous production schedules and meeting the stringent delivery timelines required by global pharmaceutical clients. The use of commercially available and stable reagents like Fmoc-amino acids and Wang resin further secures the supply chain against raw material shortages, ensuring consistent availability of the final API.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing standard solid-phase synthesis equipment that can be easily scaled from pilot plants to multi-ton commercial production. The shift towards anhydrous production conditions and the recyclability of organic solvents used for washing align with modern green chemistry principles. This reduces the volume of hazardous waste generated, simplifying waste treatment protocols and ensuring compliance with increasingly strict environmental regulations. The improved safety profile regarding solvent flash points also facilitates easier permitting and operation in diverse geographic locations, enhancing the flexibility of the global supply network.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Bivalirudin synthesis method. These answers are derived directly from the experimental data and specifications provided in patent CN101906150B, offering clarity on the feasibility and advantages of this approach for industrial application. Understanding these details is essential for technical teams evaluating the transfer of this technology to their own manufacturing facilities.
Q: How does this method reduce Bivalirudin impurities compared to traditional SPPS?
A: By pre-synthesizing difficult sequences like Gly-Gly-Gly-Gly and Asn-Gly in the liquid phase, this method eliminates the accumulation of deletion sequences (such as [Bivalirudin-Gly]) that typically occur during repetitive solid-phase coupling cycles.
Q: What specific deprotection reagents are recommended for this synthesis?
A: The patent specifies a deprotection mixture containing 3-20% piperidine and 0.5-10% DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), optionally with HOBt or HOOBT, which effectively removes Fmoc groups while minimizing side reactions.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the method utilizes standard solid-phase equipment and avoids hazardous solvents like diethyl ether in favor of MTBE, significantly improving safety profiles and environmental compliance for industrial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Bivalirudin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthesis technologies to meet the evolving demands of the pharmaceutical market. Our team of expert chemists has extensively evaluated the hybrid solid-liquid phase method described in CN101906150B and possesses the technical capability to implement this route effectively. We have extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the high purity and low impurity profiles achieved in the lab are maintained at an industrial scale. Our state-of-the-art facilities are equipped with rigorous QC labs and stringent purity specifications to guarantee that every batch of Bivalirudin meets the highest international regulatory standards.
We invite procurement leaders and R&D directors to collaborate with us to optimize their supply chains for antithrombotic agents. By leveraging our expertise in peptide synthesis and process optimization, we can help you achieve significant cost reduction in API manufacturing while ensuring a secure and reliable supply of high-purity Bivalirudin. Please contact our technical procurement team to request a Customized Cost-Saving Analysis, specific COA data, and route feasibility assessments tailored to your project requirements. Let us partner with you to bring this innovative synthesis method to commercial reality.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →