Advanced Solid-Phase Synthesis of Eptifibatide Acetate for Commercial Scale-Up
Advanced Solid-Phase Synthesis of Eptifibatide Acetate for Commercial Scale-Up
The pharmaceutical landscape for antithrombotic agents continues to evolve, driven by the demand for higher purity and more efficient manufacturing processes for critical drugs like Eptifibatide. Patent CN102584944A introduces a transformative preparation method for Eptifibatide acetate, addressing longstanding challenges in solid-phase peptide synthesis (SPPS). This innovation focuses on a strategic fragment condensation approach that bypasses the limitations of traditional stepwise amino acid addition. By utilizing specific protected amino acid fragments, namely X (Mpr-Harg) and Y (Gly-Asp), the process effectively mitigates the formation of difficult-to-remove impurities such as [+1Gly]-eptifibatide and [-1Harg]-eptifibatide. For R&D directors and procurement specialists, this represents a significant opportunity to optimize the supply chain for this vital GPIIb/IIIa receptor antagonist, ensuring both cost efficiency and regulatory compliance through superior impurity control.
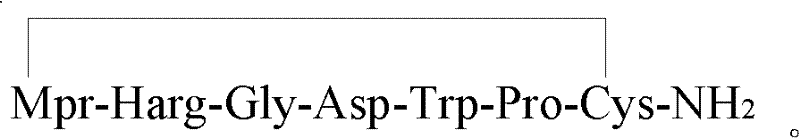
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional solid-phase synthesis of complex heptapeptides often relies on the sequential coupling of individual Fmoc-protected amino acids. While conceptually straightforward, this approach encounters severe bottlenecks when dealing with specific residue combinations found in Eptifibatide. The presence of Glycine and Homoarginine (Harg) creates a unique chemical environment prone to side reactions. Glycine, lacking a side chain, offers little steric hindrance, which can inadvertently facilitate racemization or over-coupling events leading to insertion impurities like [+1Gly]-eptifibatide. Furthermore, the bulky and complex protecting groups required for Homoarginine can lead to incomplete couplings, resulting in deletion sequences such as [-1Harg]-eptifibatide. These impurities possess polarities remarkably similar to the target molecule, making their removal via preparative HPLC extremely difficult, resource-intensive, and yield-limiting. Consequently, conventional methods often struggle to achieve the high purity standards required for parenteral administration without sacrificing overall production throughput.
The Novel Approach
The methodology disclosed in CN102584944A fundamentally restructures the synthesis strategy by introducing pre-assembled dipeptide fragments. Instead of coupling Gly and Asp individually, the process utilizes a pre-formed Fmoc-Gly-Asp(OtBu)-OH fragment. Similarly, the challenging Mpr-Harg sequence is introduced as a single Mpr(Trt)-Harg-OH unit. This fragment-based logic drastically reduces the total number of coupling cycles required on the solid support. By minimizing the number of repetitive deprotection and activation steps, the probability of generating deletion or insertion byproducts is statistically reduced. This approach not only streamlines the synthetic route but also inherently enhances the crude product profile, shifting the burden away from downstream purification and towards upstream process control. For manufacturing teams, this translates to a more robust process capable of delivering high-purity intermediates with greater consistency and reduced solvent consumption.
Mechanistic Insights into Fragment-Based Solid-Phase Peptide Synthesis
The core of this technological advancement lies in the precise orchestration of solid-phase coupling reactions using optimized resin systems. The synthesis initiates on an amino-functionalized resin, with Rink Amide MBHA resin identified as the preferred matrix due to its superior stability and loading capacity. The mechanism involves the sequential assembly of the peptide chain in a C-to-N direction, but with strategic pauses for fragment ligation. When coupling the X fragment (Mpr-Harg), the steric bulk is managed by the pre-formation of the peptide bond in solution phase, ensuring that the difficult coupling occurs under controlled homogeneous conditions rather than the heterogeneous environment of the resin bead. This ensures near-quantitative conversion. Following this, the Y fragment (Gly-Asp) is coupled, effectively bridging the gap between the hydrophobic tryptophan/proline region and the N-terminal mercaptopropionic acid. The use of potent coupling reagents like DIC (N,N'-Diisopropylcarbodiimide) in conjunction with HOBt (1-Hydroxybenzotriazole) facilitates rapid amide bond formation while suppressing racemization, a critical factor for maintaining the stereochemical integrity of the bioactive peptide.
Following the complete assembly of the resin-bound peptide, the mechanism shifts to cleavage and cyclization. The acidolysis step employs a cocktail of trifluoroacetic acid (TFA), 1,2-Ethanedithiol (EDT), and water. TFA serves as the strong acid to cleave the peptide from the resin linker and remove acid-labile side-chain protecting groups like Boc, Trt, and OtBu. EDT acts as a scavenger to prevent the alkylation of sensitive residues like Tryptophan and Cysteine by carbocations generated during cleavage. Once the linear peptide is liberated, the final critical transformation is the formation of the intramolecular disulfide bond between the thiol of Mpr and the thiol of Cys. This oxidative cyclization is typically performed using iodine in a dilute acetic acid solution. The iodine titration must be carefully monitored to ensure complete oxidation without over-oxidizing other sensitive moieties. This cyclization locks the peptide into its bioactive conformation, which is essential for its high affinity binding to the GPIIb/IIIa receptor.
How to Synthesize Eptifibatide Acetate Efficiently
The implementation of this fragment-based synthesis requires strict adherence to stoichiometric ratios and reaction times to maximize the benefits of the novel route. The process begins with the swelling of the Rink Amide MBHA resin, followed by the sequential coupling of Fmoc-Cys(Trt), Fmoc-Pro, and Fmoc-Trp(Boc). The pivotal step involves the coupling of the pre-synthesized Fmoc-Gly-Asp(OtBu)-OH fragment, which replaces two standard coupling cycles. Subsequently, the Mpr(Trt)-Harg-OH fragment is coupled to complete the sequence. Detailed operational parameters, including specific molar equivalents of activators and precise deprotection windows, are critical for reproducibility. For a comprehensive guide on executing this synthesis with maximum yield and purity, please refer to the standardized protocol outlined below.
- Load the initial amino acid onto a suitable amino resin, such as Rink Amide MBHA resin, and sequentially couple protected fragments X (Mpr-Harg) and Y (Gly-Asp) along with Trp, Pro, and Cys residues.
- Perform acidolysis on the fully assembled peptide resin using a TFA/EDT/water mixture to cleave the peptide from the resin and remove side-chain protecting groups simultaneously.
- Oxidize the resulting linear peptide crude product using iodine in acetic acid to form the critical disulfide bond between Mpr and Cys, followed by HPLC purification and salt exchange.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this fragment-based synthesis method offers tangible strategic advantages beyond mere technical elegance. The primary value driver is the substantial reduction in downstream processing costs. By significantly lowering the burden of impurities like [+1Gly] and [-1Harg] variants, the load on preparative HPLC columns is drastically reduced. This means less stationary phase is consumed, and significantly lower volumes of expensive HPLC-grade solvents are required for purification. Furthermore, the simplified coupling sequence reduces the consumption of activated amino acids and coupling reagents, directly impacting the bill of materials. The process operates under mild conditions, largely at room temperature during acidolysis, which lowers energy requirements for heating or cooling large-scale reactors. These factors combine to create a manufacturing profile that is not only more cost-effective but also more environmentally sustainable due to reduced waste generation.
- Cost Reduction in Manufacturing: The elimination of difficult-to-separate impurities directly correlates to higher recovery rates during the final purification stages. In traditional processes, a significant portion of the crude product is lost during the aggressive cutting of HPLC peaks to exclude closely eluting impurities. By improving the crude profile through fragment coupling, the yield of the final pure API is significantly enhanced. Additionally, the reduction in the total number of coupling cycles decreases the usage of expensive Fmoc-amino acids and activation reagents like HATU or PyBOP. This streamlined reagent consumption leads to a leaner cost structure per kilogram of produced Eptifibatide, allowing for more competitive pricing in the global market without compromising on quality margins.
- Enhanced Supply Chain Reliability: Reliance on complex, multi-step sequential synthesis increases the risk of batch failure; a single failed coupling in a seven-step sequence can ruin the entire batch. By condensing the synthesis into fewer, more robust fragment coupling steps, the overall process reliability is improved. The use of standard, commercially available resins like Rink Amide MBHA ensures that raw material sourcing remains stable and unaffected by niche supply constraints. Moreover, the ability to pre-synthesize and quality-control the key fragments (X and Y) before they ever touch the solid support adds an additional layer of quality assurance, ensuring that only high-quality building blocks enter the final assembly line, thereby reducing the variability in final product delivery timelines.
- Scalability and Environmental Compliance: Scaling peptide synthesis is notoriously difficult due to the swelling properties of resins and the heat generation during exothermic coupling reactions. This novel method, by reducing the number of on-resin steps, inherently simplifies the scale-up trajectory. The acidolysis step uses a standard TFA cocktail which is well-understood in industrial waste treatment facilities, facilitating easier compliance with environmental regulations. The reduction in solvent volume for purification also means a smaller carbon footprint for the manufacturing process. For large-scale production ranging from hundreds of kilograms to metric tons, this process offers a smoother path to validation and regulatory approval, as the impurity profile is cleaner and more consistent across different batch sizes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of Eptifibatide acetate using this advanced methodology. These insights are derived directly from the patent specifications and are intended to clarify the operational benefits for potential partners. Understanding these nuances is crucial for evaluating the feasibility of integrating this supply source into your existing pharmaceutical portfolio.
Q: How does the fragment coupling method improve purity compared to sequential amino acid addition?
A: By using pre-formed fragments like Mpr-Harg and Gly-Asp, the method significantly reduces the number of coupling cycles. This minimizes the formation of deletion sequences (e.g., [-1Harg]) and insertion impurities (e.g., [+1Gly]) that are common when coupling sensitive residues like Glycine and Homoarginine individually.
Q: What is the preferred resin system for this synthesis?
A: The patent identifies Rink Amide MBHA resin as the preferred solid support due to its optimal swelling properties and compatibility with the acidolysis conditions required to release the C-terminal amide of Eptifibatide.
Q: How is the cyclic structure of Eptifibatide formed?
A: The cyclic structure is established through an oxidative cyclization step where the thiol groups of the Mercaptopropionic acid (Mpr) and Cysteine (Cys) residues are oxidized to form a disulfide bond, typically using iodine as the oxidizing agent.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Eptifibatide Acetate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and robust manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated fragment-coupling chemistry described in CN102584944A can be executed flawlessly at an industrial level. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify that every batch of Eptifibatide acetate meets the >99.5% purity benchmark. Our commitment to quality ensures that the complex impurity profiles associated with traditional synthesis are kept well below regulatory thresholds, safeguarding your drug development timeline.
We invite you to collaborate with us to leverage this advanced synthesis technology for your supply chain. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis that quantifies the potential efficiencies of switching to this fragment-based route. We encourage you to contact us to obtain specific COA data from our pilot batches and to discuss detailed route feasibility assessments tailored to your volume requirements. Let us help you secure a reliable, high-quality supply of this critical cardiovascular therapeutic.