Advanced Solid-Phase Synthesis of Eptifibatide Acetate for Commercial Scalability and High Purity
Advanced Solid-Phase Synthesis of Eptifibatide Acetate for Commercial Scalability and High Purity
The pharmaceutical industry continuously seeks robust manufacturing pathways for complex polypeptides that balance high purity with economic feasibility. Patent CN102584944A introduces a significant technological advancement in the preparation of Eptifibatide acetate, a potent platelet glycoprotein IIb/IIIa receptor antagonist used in acute coronary syndromes. This invention addresses critical bottlenecks in traditional solid-phase peptide synthesis (SPPS) by employing a novel fragment condensation strategy that drastically minimizes the generation of difficult-to-remove impurities. By shifting from a purely stepwise amino acid addition to a hybrid approach utilizing protected dipeptide fragments, the process achieves a finished product purity of greater than 99.5%, setting a new benchmark for quality in antiplatelet therapy manufacturing. This technical breakthrough not only enhances the safety profile of the medication by reducing impurity loads but also streamlines the downstream purification workflow, offering substantial value for contract development and manufacturing organizations (CDMOs) aiming to optimize their peptide production lines.
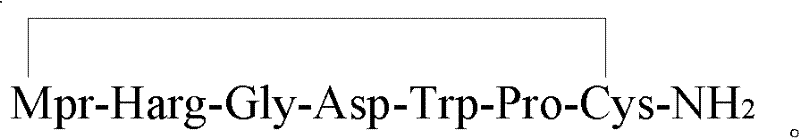
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional methods for synthesizing Eptifibatide often rely on the sequential inoculation of single protected amino acids onto a solid support, a process that becomes increasingly problematic as the peptide chain elongates. Specifically, the presence of Glycine (Gly) and the structural characteristics of the protected Homoarginine (Harg) residue create a high propensity for side reactions during the coupling phases. In standard Fmoc solid-phase synthesis, these specific residues are prone to generating deletion sequences such as [-1Gly]-Eptifibatide and [-1Harg]-Eptifibatide, as well as insertion impurities like [+1Gly]-Eptifibatide. These structurally similar byproducts possess polarities that closely mimic the target molecule, thereby exponentially increasing the difficulty of purification via preparative high-performance liquid chromatography (HPLC). The accumulation of these impurities not only depresses the overall yield of the crude product but also necessitates aggressive and costly purification steps that can degrade the final active pharmaceutical ingredient, ultimately compromising the economic viability and supply reliability of the manufacturing process.
The Novel Approach
The methodology disclosed in patent CN102584944A circumvents these inherent limitations by introducing a strategic fragmentation of the peptide sequence into distinct, pre-activated building blocks. Instead of coupling individual amino acids for the entire sequence, the invention utilizes specific protected fragments, designated as Fragment X (Mpr-Harg) and Fragment Y (Gly-Asp), which are ligated onto the growing peptide chain in fewer, more controlled steps. This fragment-based approach effectively bypasses the problematic coupling environments that typically lead to Glycine insertion or Harg deletion, thereby directly suppressing the formation of the notorious [+1Gly] and [-1Harg] impurities at the source. By reducing the total number of coupling cycles required for the critical regions of the peptide, the novel approach significantly enhances the reaction efficiency and ensures a much cleaner crude product profile. This structural simplification of the synthesis route translates directly into operational excellence, allowing manufacturers to achieve higher throughput with reduced solvent consumption and reagent waste, aligning perfectly with modern green chemistry principles and cost-reduction mandates.
Mechanistic Insights into Fragment-Based Solid-Phase Peptide Synthesis
The core mechanistic advantage of this process lies in the precise engineering of the coupling segments to sterically and electronically favor the formation of the desired peptide bonds while minimizing racemization and deletion. Fragment X, comprising Mercaptopropionic acid (Mpr) and Homoarginine (Harg), and Fragment Y, comprising Glycine and Aspartic Acid (Asp), are synthesized with orthogonal protecting groups such as Trt, Acm, Pbf, and OtBu to ensure selective deprotection. During the solid-phase assembly on the amino resin, these fragments are activated using efficient condensing agents like DIC (N,N'-Diisopropylcarbodiimide) in the presence of additives like HOBt (1-Hydroxybenzotriazole) to form active esters that react rapidly with the free amine groups on the resin-bound peptide. The use of Rink Amide MBHA resin, which is highlighted as the preferred solid support, provides an optimal loading capacity (0.5-1.0 mmol/g) that balances reaction kinetics with steric accessibility, ensuring that the bulky fragments can couple efficiently without causing aggregation or incomplete reactions that would otherwise lead to truncated sequences.
Following the assembly of the full linear sequence on the solid support, the mechanism shifts to a simultaneous cleavage and deprotection strategy using a carefully optimized acidolysis cocktail. The patent specifies a mixture of Trifluoroacetic acid (TFA), 1,2-Ethanedithiol (EDT), and water, typically in a ratio of roughly 90:5:5, which serves a dual purpose: cleaving the peptide from the resin linker and removing acid-labile side-chain protecting groups like Boc, Trt, and Pbf. The inclusion of EDT is critical as it acts as a scavenger to prevent the alkylation of sensitive residues like Tryptophan and Methionine by carbocation byproducts generated during acidolysis. Once the linear peptide is liberated into solution, the final mechanistic step involves the oxidative cyclization of the thiol groups on the Mpr and Cysteine (Cys) residues to form the essential disulfide bridge. This oxidation is meticulously controlled using iodine titration in an acetic acid medium, ensuring the formation of the correct cyclic heptapeptide structure without over-oxidation to sulfones or sulfonic acids, which would render the batch unusable.
How to Synthesize Eptifibatide Acetate Efficiently
The synthesis of Eptifibatide acetate via this patented route requires strict adherence to the defined fragment coupling protocols and purification parameters to ensure the high purity specifications are met consistently. The process begins with the swelling of the preferred Rink Amide MBHA resin, followed by the sequential coupling of the protected fragments and single amino acids using standard Fmoc chemistry protocols with DIC/HOBt activation. Critical attention must be paid to the coupling times, which are optimized between 100 to 140 minutes to ensure completeness without excessive exposure to activation reagents that could induce side reactions. After the full sequence is assembled, the resin is treated with the TFA/EDT/water cleavage mixture for approximately 2 hours at room temperature to release the linear peptide, which is then precipitated in cold ether and dried.
- Perform solid-phase coupling using specific protected fragments X (Mpr-Harg) and Y (Gly-Asp) on amino resin to form the peptide chain.
- Execute acidolysis using a TFA/EDT/Water mixture to cleave the peptide from the resin and remove side-chain protecting groups simultaneously.
- Oxidize the linear peptide crude product using iodine to form the critical disulfide bond, followed by HPLC purification and salt exchange.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this fragment-based synthesis method offers compelling advantages that extend far beyond simple chemical yield improvements. By fundamentally altering the synthesis strategy to avoid the formation of hard-to-separate impurities, the process eliminates the need for extensive and repetitive purification cycles that typically consume vast amounts of expensive HPLC columns and organic solvents. This reduction in downstream processing complexity translates directly into a significant reduction in manufacturing costs, as the facility can process larger batch sizes in less time with lower utility and consumable expenditures. Furthermore, the robustness of the fragment coupling method enhances supply chain reliability by reducing the risk of batch failures due to out-of-specification impurity profiles, ensuring a more consistent and predictable delivery schedule for downstream formulation teams who depend on timely API availability for drug product manufacturing.
- Cost Reduction in Manufacturing: The elimination of difficult-to-remove impurities such as [+1Gly] and [-1Harg] variants means that the crude product entering the purification stage is of substantially higher quality, thereby reducing the load on preparative chromatography systems. This efficiency gain allows for the use of smaller column volumes or fewer purification runs to achieve the same final purity, leading to drastic savings in stationary phase costs and solvent disposal fees. Additionally, the use of pre-formed fragments reduces the total number of coupling steps, which lowers the consumption of expensive activating reagents and protected amino acids, further driving down the raw material cost per kilogram of the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: Traditional peptide synthesis is often plagued by variability in crude quality, which can lead to unpredictable purification timelines and potential delays in product release. By implementing a synthesis route that inherently suppresses the formation of critical impurities, manufacturers can achieve a much more consistent production cycle time, mitigating the risk of supply disruptions caused by failed batches or extended reprocessing. This stability is crucial for maintaining continuous supply to pharmaceutical partners, especially for critical care medications like Eptifibatide where inventory buffers must be maintained to meet hospital demand without interruption.
- Scalability and Environmental Compliance: The simplified operational protocol, characterized by fewer reaction steps and milder conditions, facilitates easier scale-up from pilot plant to commercial production scales without the need for complex engineering modifications. The reduction in solvent usage and chemical waste generation associated with the streamlined purification process also aligns with increasingly stringent environmental regulations, reducing the carbon footprint of the manufacturing site and lowering the costs associated with waste treatment and regulatory compliance reporting.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this advanced synthesis technology, providing clarity on its operational benefits and quality outcomes. These insights are derived directly from the experimental data and technical specifications outlined in the patent documentation, ensuring that stakeholders have accurate information for decision-making. Understanding these details is essential for evaluating the feasibility of integrating this method into existing manufacturing portfolios or for sourcing high-quality intermediates from specialized suppliers.
Q: How does this method improve purity compared to traditional stepwise synthesis?
A: By using pre-formed fragments X and Y instead of single amino acid coupling for critical sequences, the method effectively avoids the formation of deletion and insertion impurities such as [+1Gly] and [-1Harg] eptifibatide, resulting in crude purity exceeding 99.5%.
Q: What resin is preferred for this solid-phase synthesis process?
A: While various amino resins like Rink Amide and Sieber resin can be used, the patent specifically identifies Rink Amide MBHA resin as the preferred carrier due to its optimal substitution value and compatibility with the fragment coupling conditions.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the method simplifies the operation by reducing the number of coupling cycles and purification difficulties, making it highly beneficial for realizing large-scale solid-phase synthesis processes with consistent quality control.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Eptifibatide Acetate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic methodologies to meet the rigorous demands of the global pharmaceutical market. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless and efficient. We are committed to delivering high-purity Eptifibatide acetate that meets stringent purity specifications through our rigorous QC labs, which utilize state-of-the-art analytical instrumentation to verify every batch against the highest international standards. Our dedication to technical excellence ensures that our clients receive a product that is not only chemically pure but also manufactured with a level of consistency that supports their own regulatory filings and commercial success.
We invite potential partners to engage with our technical procurement team to discuss how our capabilities can support your specific supply chain needs. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of how our optimized synthesis routes can reduce your overall cost of goods sold while improving supply security. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project requirements, allowing you to make informed decisions that drive value and efficiency in your pharmaceutical development pipeline.