Advanced Synthesis of Bempedoic Acid Intermediates: A Scalable Industrial Solution
Introduction to Advanced Bempedoic Acid Synthesis
The landscape of cardiovascular drug manufacturing is undergoing a significant transformation with the emergence of novel synthetic pathways for key intermediates. Patent CN111170855A introduces a groundbreaking method for synthesizing 8-hydroxy-2,2,14,14-tetramethylpentadecanedioic acid, widely known as Bempedoic Acid. This compound serves as a potent ATP Citrate Lyase (ACL) inhibitor, offering a crucial therapeutic alternative for patients with dyslipidemia who cannot tolerate statins. The structural complexity of this molecule, characterized by its long carbon chain and specific methyl substitutions, has historically posed challenges for efficient industrial production. 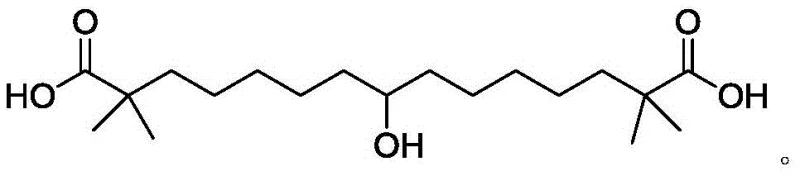 . The disclosed technology addresses these challenges by providing a route that is not only chemically robust but also economically viable for large-scale operations, marking a pivotal shift from laboratory-scale curiosity to industrial reality.
. The disclosed technology addresses these challenges by providing a route that is not only chemically robust but also economically viable for large-scale operations, marking a pivotal shift from laboratory-scale curiosity to industrial reality.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as the route reported in WO2004067489, have long been the standard but suffer from severe drawbacks that hinder modern pharmaceutical manufacturing. 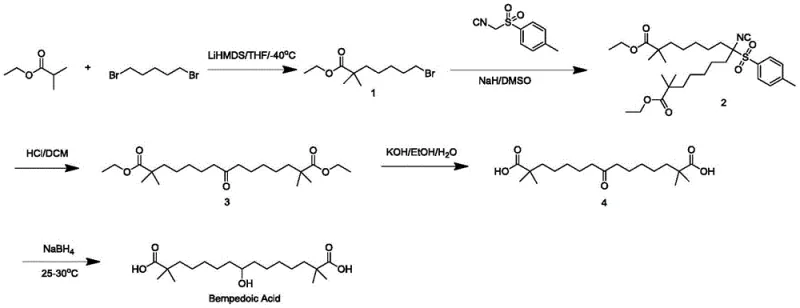 . These traditional processes rely heavily on the use of p-toluenesulfonyl methyl isocyanide, a reagent known for its high toxicity and poor atom economy, which creates substantial waste disposal issues. Furthermore, the reliance on sodium hydride as a strong base introduces significant safety risks due to its pyrophoric nature, requiring specialized handling equipment and inert atmospheres that drive up capital expenditure. Perhaps most critically for quality control, these older routes generate potential genotoxic impurities, specifically p-toluenesulfonyl derivatives, which are notoriously difficult to remove and pose regulatory hurdles for API approval. The necessity for column chromatography purification in intermediate steps further exacerbates cost and throughput limitations, rendering these methods unsuitable for the high-volume demands of the global lipid-lowering drug market.
. These traditional processes rely heavily on the use of p-toluenesulfonyl methyl isocyanide, a reagent known for its high toxicity and poor atom economy, which creates substantial waste disposal issues. Furthermore, the reliance on sodium hydride as a strong base introduces significant safety risks due to its pyrophoric nature, requiring specialized handling equipment and inert atmospheres that drive up capital expenditure. Perhaps most critically for quality control, these older routes generate potential genotoxic impurities, specifically p-toluenesulfonyl derivatives, which are notoriously difficult to remove and pose regulatory hurdles for API approval. The necessity for column chromatography purification in intermediate steps further exacerbates cost and throughput limitations, rendering these methods unsuitable for the high-volume demands of the global lipid-lowering drug market.
The Novel Approach
In stark contrast, the methodology outlined in CN111170855A presents a streamlined, safer, and more efficient synthetic strategy that bypasses the pitfalls of its predecessors. 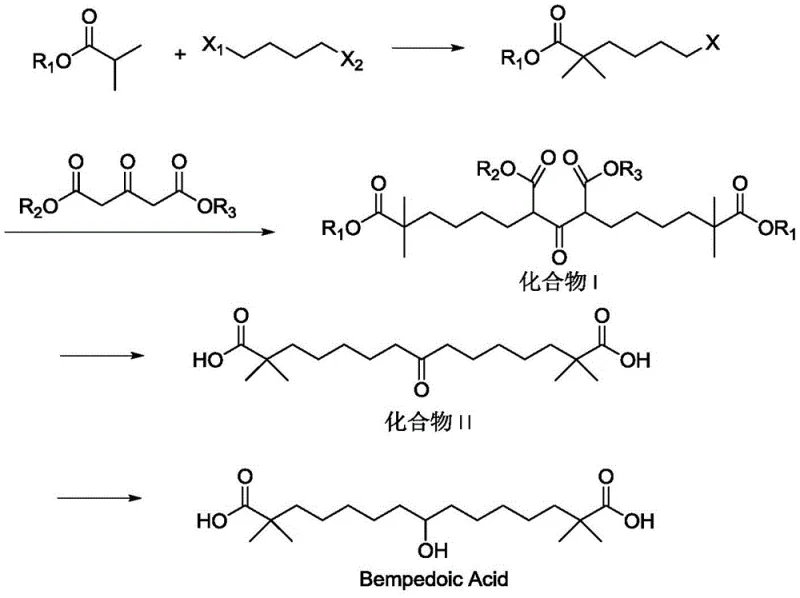 . This innovative approach utilizes a condensation reaction between 6-halo-2,2-dimethylhexanoate and acetone dicarboxylic acid diester, catalyzed by benign alkali metal carbonates rather than hazardous hydrides. By eliminating the toxic isocyanide reagent entirely, the process achieves superior atom economy and drastically reduces the environmental footprint of the synthesis. The route is designed with scalability in mind, replacing complex chromatographic purifications with straightforward crystallization techniques that are easily adaptable to multi-ton reactors. This shift not only enhances the safety profile for plant operators but also ensures a cleaner impurity profile, directly addressing the stringent quality requirements of regulatory bodies for cardiovascular medications.
. This innovative approach utilizes a condensation reaction between 6-halo-2,2-dimethylhexanoate and acetone dicarboxylic acid diester, catalyzed by benign alkali metal carbonates rather than hazardous hydrides. By eliminating the toxic isocyanide reagent entirely, the process achieves superior atom economy and drastically reduces the environmental footprint of the synthesis. The route is designed with scalability in mind, replacing complex chromatographic purifications with straightforward crystallization techniques that are easily adaptable to multi-ton reactors. This shift not only enhances the safety profile for plant operators but also ensures a cleaner impurity profile, directly addressing the stringent quality requirements of regulatory bodies for cardiovascular medications.
Mechanistic Insights into Alkylation and Condensation Chemistry
The core of this synthetic breakthrough lies in the precise control of alkylation and subsequent condensation reactions. The process initiates with the alkylation of isobutyrate esters using 1,4-dihalobutane in the presence of a non-nucleophilic base such as lithium diisopropylamide (LiHMDS) or sodium bis(trimethylsilyl)amide. 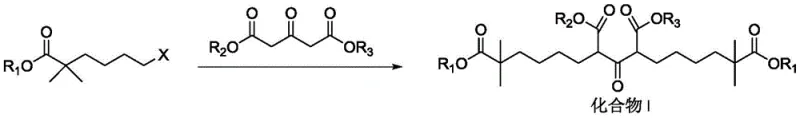 . This step is critical for establishing the gem-dimethyl motif at the 2-position of the hexanoate chain, a structural feature essential for the biological activity of the final API. The reaction conditions are meticulously optimized, typically operating at temperatures between -70°C and 30°C, to minimize side reactions such as elimination or over-alkylation. Following the formation of the halo-ester intermediate, the process employs a double alkylation strategy using dialkyl acetone dicarboxylate. This condensation step, facilitated by potassium carbonate and potassium iodide in polar aprotic solvents, effectively stitches together the two halves of the carbon skeleton. The use of potassium iodide acts as a catalyst to enhance the nucleophilicity of the halide leaving group, ensuring high conversion rates even at moderate temperatures of 30-80°C.
. This step is critical for establishing the gem-dimethyl motif at the 2-position of the hexanoate chain, a structural feature essential for the biological activity of the final API. The reaction conditions are meticulously optimized, typically operating at temperatures between -70°C and 30°C, to minimize side reactions such as elimination or over-alkylation. Following the formation of the halo-ester intermediate, the process employs a double alkylation strategy using dialkyl acetone dicarboxylate. This condensation step, facilitated by potassium carbonate and potassium iodide in polar aprotic solvents, effectively stitches together the two halves of the carbon skeleton. The use of potassium iodide acts as a catalyst to enhance the nucleophilicity of the halide leaving group, ensuring high conversion rates even at moderate temperatures of 30-80°C.
Impurity control is inherently built into the mechanistic design of this route. Unlike previous methods that generate sulfonamide byproducts, the primary byproducts here are simple inorganic salts and unreacted starting materials that are easily removed during the aqueous workup. The hydrolysis step, which converts the tri-ester intermediate into the dicarboxylic acid, is performed under controlled alkaline or acidic conditions at elevated temperatures (60-100°C). This thermal energy drives the decarboxylation of the central acetone moiety, yielding the 8-keto intermediate with high fidelity. The final reduction of the ketone to the secondary alcohol is achieved using sodium borohydride, a selective reducing agent that leaves the ester and carboxylic acid functionalities intact. This chemoselectivity is paramount for maintaining the integrity of the molecule while introducing the crucial 8-hydroxy group, resulting in a final product with minimal structural analogs or stereoisomeric impurities.
How to Synthesize 8-Hydroxy-2,2,14,14-tetramethylpentadecanedioic Acid Efficiently
The execution of this synthesis requires careful attention to stoichiometry and temperature control to maximize yield and purity. The process begins with the preparation of the halo-ester precursor, followed by the key condensation with acetone dicarboxylate, hydrolysis, and final reduction. Each step has been optimized to balance reaction kinetics with operational simplicity, ensuring that the protocol is robust enough for transfer from the laboratory to the production floor. For R&D teams looking to implement this technology, understanding the nuances of the workup procedures—particularly the pH adjustments and solvent choices for crystallization—is essential for replicating the high purity levels reported in the patent data.
- Alkylation of isobutyrate with 1,4-dihalobutane using LiHMDS or NaHMDS base at low temperature (-70 to 30°C) to form 6-halo-2,2-dimethylhexanoate.
- Condensation of the halo-ester with dialkyl acetone dicarboxylate in the presence of potassium carbonate and potassium iodide at 30-80°C.
- Hydrolysis of the condensation product under alkaline or acidic conditions followed by acidification to obtain 8-keto-2,2,14,14-tetramethylpentadecanedioic acid.
- Reduction of the keto-diacid using sodium borohydride or similar reducing agents in aqueous or alcoholic solvent systems to yield the final hydroxy-diacid.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route offers compelling strategic advantages that extend beyond mere technical feasibility. The elimination of hazardous reagents like sodium hydride and toxic isocyanides translates directly into reduced safety compliance costs and lower insurance premiums for manufacturing facilities. Furthermore, the avoidance of column chromatography, a batch process that is notoriously difficult to scale and solvent-intensive, allows for a continuous or semi-continuous manufacturing flow. This operational efficiency significantly reduces the consumption of organic solvents, aligning with green chemistry principles and lowering waste disposal expenses. The ability to purify intermediates and the final product through crystallization rather than chromatography ensures a more consistent supply of high-quality material, mitigating the risk of batch failures that can disrupt the entire production schedule.
- Cost Reduction in Manufacturing: The streamlined nature of this process eliminates several costly unit operations found in traditional routes. By removing the need for expensive and dangerous reagents, the raw material costs are substantially lowered. Additionally, the replacement of chromatographic purification with crystallization drastically reduces solvent usage and processing time, leading to significant operational expenditure savings. The higher atom economy of the condensation step means less waste is generated per kilogram of product, further enhancing the overall cost-efficiency of the manufacturing campaign.
- Enhanced Supply Chain Reliability: The reliance on commercially available and stable starting materials, such as isobutyrate esters and dihaloalkanes, ensures a robust supply chain that is less susceptible to market volatility. The simplified process flow reduces the number of intermediate isolation steps, thereby shortening the overall lead time from raw material intake to finished API intermediate. This agility allows manufacturers to respond more quickly to fluctuations in market demand for lipid-lowering therapies, ensuring a steady flow of material to downstream formulation partners without the bottlenecks associated with complex purification protocols.
- Scalability and Environmental Compliance: The mild reaction conditions and the use of standard industrial reagents make this process highly scalable from pilot plant to commercial production volumes. The absence of genotoxic impurities simplifies the regulatory filing process, reducing the time to market for generic versions of the drug. Moreover, the reduced solvent load and safer reagent profile facilitate easier compliance with increasingly stringent environmental regulations, positioning manufacturers as responsible stewards of sustainable chemical production practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing a clear picture of what stakeholders can expect during technology transfer.
Q: How does the new synthesis route improve safety compared to prior art?
A: The novel route described in CN111170855A eliminates the use of highly toxic p-toluenesulfonyl methyl isocyanide and dangerous sodium hydride, replacing them with safer alkali metal carbonates and standard reducing agents, significantly reducing operational hazards.
Q: What are the purity advantages of this method for API manufacturing?
A: By avoiding the generation of potential genotoxic impurities like p-toluenesulfonic acid derivatives and utilizing crystallization instead of column chromatography for purification, this method ensures higher product purity suitable for strict pharmaceutical standards.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process features short reaction steps, mild conditions (30-80°C), and avoids complex purification techniques like column chromatography, making it highly amenable to scale-up from pilot plant to multi-ton commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Bempedoic Acid Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic routes to maintain competitiveness in the global pharmaceutical market. Our team of expert chemists has extensively evaluated the methodology described in CN111170855A and possesses the technical capability to scale this diverse pathway from 100 kgs to 100 MT/annual commercial production. We are committed to delivering high-purity intermediates that meet stringent purity specifications, leveraging our rigorous QC labs to ensure every batch complies with international pharmacopoeia standards. Our facility is equipped to handle the specific solvent systems and temperature controls required for this synthesis, guaranteeing a reliable supply of this vital cardiovascular drug intermediate.
We invite potential partners to engage with our technical procurement team to discuss how this optimized route can benefit your specific supply chain needs. By requesting a Customized Cost-Saving Analysis, you can gain a detailed understanding of the economic impact of switching to this greener, more efficient process. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project timelines, ensuring a seamless transition to a more sustainable and cost-effective manufacturing strategy.