Advanced Asymmetric Synthesis of Chiral Statin Side Chains for Commercial Scale-Up
Introduction to Next-Generation Statin Intermediate Technology
The global demand for high-efficacy lipid-lowering agents continues to drive innovation in the synthesis of complex pharmaceutical intermediates. Patent CN102212081A discloses a groundbreaking preparation method for a key chiral intermediate, specifically (3R)-3-tertbutyldimethylsiloxy-6-dialkoxyphosphono-5-oxo-tertbutyl hexanate, often referred to as Formula 1. This compound serves as a critical precursor for the synthesis of super-statins such as Rosuvastatin and Pitavastatin, which are renowned for their potent HMG-CoA reductase inhibitory activity. The structural complexity of these molecules, featuring multiple chiral centers and a trans-double bond, presents significant synthetic challenges that this new methodology aims to overcome.
Traditional approaches to constructing the statin side chain have often relied on resolution strategies that are inherently inefficient. In contrast, the technology outlined in CN102212081A employs a sophisticated asymmetric induction strategy. By leveraging chiral auxiliaries early in the synthetic sequence, the process ensures high optical purity without the material losses typical of racemic separation. This advancement represents a significant leap forward for any reliable pharmaceutical intermediate supplier seeking to optimize production workflows for high-value cardiovascular therapeutics.
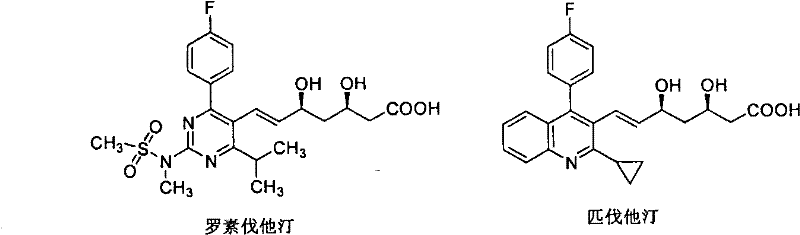
The relevance of this intermediate extends beyond mere academic interest; it is the backbone of some of the most commercially successful drugs in the cardiovascular sector. As shown in the structural diagrams, the side chain containing the chiral hydroxy groups and the phosphonate moiety is essential for biological activity. Mastering the synthesis of this fragment with high fidelity is paramount for ensuring the efficacy and safety of the final API. This patent provides a robust roadmap for achieving exactly that, addressing the long-standing difficulties associated with statin side chain construction.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those disclosed in Document WO 2008130678, have historically relied on the resolution of racemic mixtures to obtain the desired chiral configuration. A common strategy involves synthesizing a racemic glutarate derivative and subsequently separating the enantiomers using chiral amines, such as phenylethylamine. While chemically feasible, this approach suffers from a fundamental thermodynamic limitation: the maximum theoretical yield for the desired enantiomer is capped at 50%. The unwanted enantiomer is typically discarded or requires costly recycling processes, leading to substantial material waste and increased production costs.
Furthermore, conventional routes often utilize methyl esters as protecting groups for the carboxylic acid functionality. As noted in the background technology of the patent, methyl esters can be prone to side reactions under basic conditions, potentially leading to the formation of lactone intermediates. This lactonization can cause racemization at the chiral center, thereby compromising the optical purity of the final product. Such impurities are unacceptable in the manufacture of high-purity OLED material or pharmaceutical grades, where strict regulatory standards mandate exceptional enantiomeric excess. The reliance on resolution also introduces additional unit operations, such as repeated crystallizations, which complicate the process flow and extend lead times.
The Novel Approach
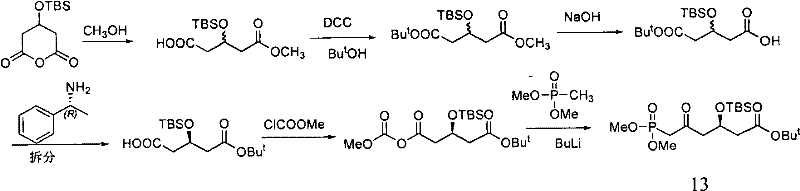
The method disclosed in CN102212081A fundamentally shifts the paradigm from separation to induction. Instead of creating a racemate and trying to fix it later, the process installs the chiral center at the very beginning using S-benzyl mandelate. This chiral auxiliary directs the stereochemistry of the subsequent nucleophilic attack on the glutaric anhydride derivative. This strategic change eliminates the need for resolution entirely, theoretically allowing for 100% conversion of starting materials into the desired chiral framework. This efficiency is a game-changer for cost reduction in statin manufacturing, as it maximizes atom economy and minimizes waste generation.
Additionally, the novel route employs a tert-butyl ester protecting group rather than a methyl ester. The patent explicitly highlights that the tert-butyl group offers superior stability against base-catalyzed hydrolysis and lactonization compared to its methyl counterpart. This stability ensures that the chiral integrity of the molecule is maintained throughout the harsh conditions of subsequent synthetic steps, such as the Wittig-Horner reaction. By combining asymmetric induction with robust protecting group chemistry, this approach delivers a streamlined, high-yielding pathway that is ideally suited for the commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Asymmetric Induction and Phosphonate Coupling
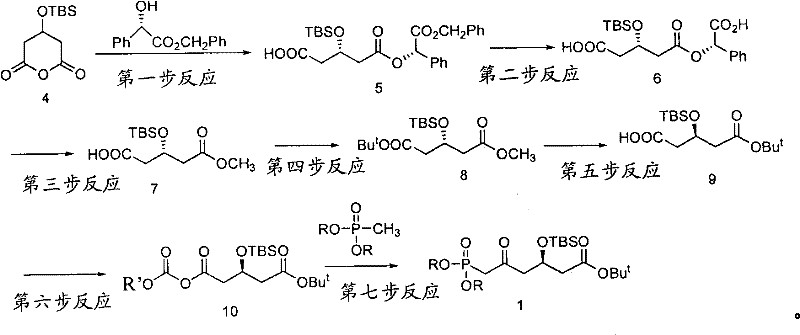
The core of this synthetic innovation lies in the first step, where stereochemical control is established. The reaction involves the generation of a lithiated enolate from S-benzyl mandelate using a strong base like n-Butyl Lithium at cryogenic temperatures, specifically around -78°C. This enolate then attacks the carbonyl carbon of 3-(tert-butyldimethylsilyl)-glutaric anhydride. The steric environment created by the bulky silyl group and the chiral mandelate backbone dictates the facial selectivity of this attack, ensuring the formation of the (3S)-configured intermediate with high diastereoselectivity. This precise control at the molecular level is what allows the process to bypass the need for downstream purification of enantiomers.
Following the establishment of chirality, the synthesis proceeds through a series of functional group transformations designed to preserve this stereochemical information. The removal of the benzyl group via catalytic hydrogenation using Pd/C is a clean and efficient step that reveals the free carboxylic acid without affecting the silyl ether or the newly formed chiral center. Subsequent steps involve transesterification to swap the benzyl ester for a methyl ester, followed by condensation with tert-butanol to install the stable tert-butyl ester. The final critical transformation is the introduction of the phosphonate group via a mixed anhydride method. Reacting the carboxylic acid with methyl chloroformate generates a reactive intermediate that is immediately attacked by the anion of dimethyl methylphosphonate, generated in situ using n-Butyl Lithium. This sequence constructs the beta-keto phosphonate motif essential for the final Wittig-Horner olefination.
Impurity control is meticulously managed through the choice of reagents and conditions. For instance, the use of tert-butyl esters prevents the formation of lactones that could scramble chirality. Furthermore, the low-temperature conditions employed during the lithiation and phosphonate addition steps minimize thermal degradation and side reactions such as self-condensation. The patent reports that the intermediate obtained after hydrogenation can achieve an optical purity (ee%) exceeding 99.5% after simple recrystallization. This level of purity is critical for R&D directors who must ensure that the impurity profile of the API meets stringent international pharmacopoeia standards. The mechanism effectively decouples the complexity of chiral synthesis from the bulk manufacturing process, making high quality accessible at scale.
How to Synthesize (3R)-3-tertbutyldimethylsiloxy-6-dialkoxyphosphono-5-oxo-tertbutyl hexanate Efficiently
The synthesis of this high-value intermediate is achieved through a logical seven-step sequence that prioritizes yield and stereochemical integrity. The process begins with readily available starting materials and utilizes standard organic transformations that are well-understood in industrial settings. From the initial asymmetric alkylation to the final phosphonate coupling, each step has been optimized to minimize handling and maximize throughput. The detailed standardized synthesis steps below outline the specific conditions, reagents, and workup procedures required to replicate this high-efficiency route in a production environment.
- React 3-(tert-butyldimethylsilyl)-glutaric anhydride with S-benzyl mandelate using n-Butyl Lithium at -78°C to form the chiral diester intermediate.
- Perform catalytic hydrogenation using Pd/C to remove the benzyl protecting group, yielding the mono-acid intermediate with high optical purity.
- Execute transesterification with sodium methylate, followed by DCC-mediated condensation with tert-butanol to protect the carboxyl group.
- Hydrolyze the methyl ester selectively using sodium hydroxide, then activate with methyl chloroformate before reacting with dimethyl methylphosphonate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the shift from resolution-based synthesis to asymmetric induction offers profound economic and logistical benefits. The most immediate impact is the drastic improvement in material efficiency. By eliminating the 50% yield loss inherent in resolving racemates, the overall consumption of raw materials is significantly reduced. This reduction translates directly into lower variable costs per kilogram of the intermediate. Moreover, the avoidance of chiral resolving agents, which can be expensive and difficult to source in bulk, further simplifies the supply chain and reduces dependency on niche chemical vendors.
- Cost Reduction in Manufacturing: The elimination of resolution steps removes the need for multiple crystallization cycles and the disposal of unwanted enantiomers. This streamlining of the process reduces solvent usage, energy consumption for heating and cooling cycles, and labor hours associated with extended purification. Additionally, the use of stable tert-butyl protecting groups reduces the risk of batch failures due to racemization, ensuring consistent first-pass yields. These factors combine to create a substantially more cost-effective manufacturing process compared to legacy methods described in documents like US 526440.
- Enhanced Supply Chain Reliability: The reagents used in this novel pathway, such as S-benzyl mandelate, n-Butyl Lithium, and tert-butanol, are commodity chemicals available from multiple global suppliers. This diversification of the supply base mitigates the risk of shortages that can occur with specialized chiral catalysts or resolving agents. The robustness of the chemistry also means that the process is less sensitive to minor variations in raw material quality, ensuring consistent production schedules. For supply chain planners, this reliability is crucial for maintaining continuous API production and meeting market demand without interruption.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing reactions that translate well from the laboratory to multi-ton reactors. The absence of heavy metal catalysts (other than recyclable Pd/C) and the reduction in waste streams align with modern green chemistry principles. This environmental compatibility simplifies regulatory compliance and waste treatment costs. The ability to scale up complex chiral intermediates without exponential increases in cost or complexity makes this technology an attractive option for long-term strategic sourcing of statin precursors.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and comparative analysis provided in the patent documentation. Understanding these nuances is essential for technical teams evaluating the feasibility of adopting this route for their own manufacturing pipelines.
Q: How does this method improve optical purity compared to resolution methods?
A: By utilizing S-benzyl mandelate as a chiral auxiliary in the initial nucleophilic attack, the method induces chirality directly rather than separating racemates. This avoids the inherent 50% yield loss associated with traditional resolution techniques described in prior art like WO 2008130678.
Q: What are the critical temperature controls required for the phosphonate addition?
A: The final step involving the reaction of the mixed anhydride with dialkyl methyl phosphonate requires strict low-temperature control, specifically between -100°C and 0°C, with a preferred operating range of -78°C to prevent side reactions and ensure high regioselectivity.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the process eliminates complex purification steps like chiral chromatography or repeated recrystallizations needed for resolution. The use of standard reagents like n-Butyl Lithium and Pd/C hydrogenation makes it highly adaptable for multi-kilogram to ton-scale production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Statin Intermediate Supplier
The technological advancements detailed in CN102212081A underscore the complexity and sophistication required to produce high-quality statin intermediates. At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to bring such advanced synthetic routes to life. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. We operate stringent purity specifications and maintain rigorous QC labs to guarantee that every batch of intermediate meets the highest global standards for pharmaceutical applications.
We invite you to collaborate with us to leverage this cutting-edge synthesis technology for your statin projects. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our commitment to innovation can drive value and efficiency in your supply chain.