Advanced Industrial Synthesis of Sorafenib Intermediate: Technical & Commercial Analysis
Advanced Industrial Synthesis of Sorafenib Intermediate: Technical & Commercial Analysis
The pharmaceutical industry constantly seeks robust, scalable, and cost-effective routes for critical oncology intermediates. Patent CN101052619A presents a significant technological advancement in the preparation of 4-{4-[({[4-chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-N-methylpyridine-2-carboxamide and its tosylate salt, a key precursor in the synthesis of Raf kinase inhibitors like Sorafenib. This patent addresses the limitations of earlier methods described in WO 00/42012 and WO 03/068228 by optimizing reaction conditions, solvent systems, and purification protocols to meet rigorous GMP standards for industrial production ranging from kilograms to metric tons. The disclosed methodology focuses on enhancing purity, environmental compatibility, and volumetric yield, making it a highly attractive route for commercial manufacturing.
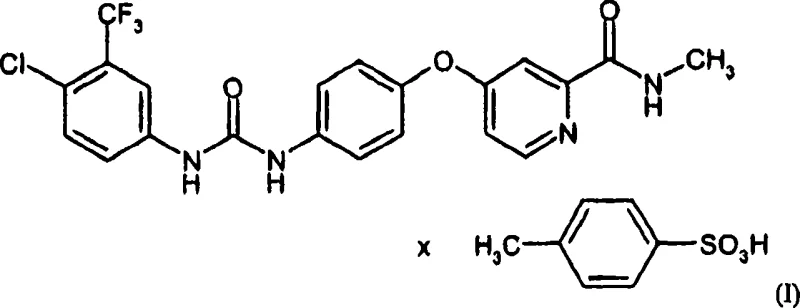
The core innovation lies in a multi-step synthetic pathway that begins with the activation of 2-pyridinecarboxylic acid and culminates in a sophisticated crystallization of the final tosylate salt. By replacing hazardous reagents and optimizing phase transfer conditions, the process ensures a consistent supply of high-purity material essential for downstream API synthesis. For procurement and supply chain leaders, understanding these technical nuances is vital for evaluating long-term vendor reliability and cost structures in the competitive landscape of pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods, such as those detailed by Bankston et al. and in WO 00/42012, typically rely on a sequence involving the formation of an acid chloride using thionyl chloride and dimethylformamide (DMF), followed by amidation and etherification steps that often require harsh conditions. A major drawback of these conventional routes is the reliance on dichloromethane (DCM) for the final urea formation step, which poses significant environmental and regulatory challenges due to its classification as a hazardous air pollutant. Furthermore, the traditional etherification step often necessitates the use of potassium carbonate and extensive extraction procedures to remove inorganic salts, leading to lower volumetric yields and increased waste generation. The formation of the acid chloride using DMF also generates dimethylcarbamoyl chloride, a corrosive byproduct that complicates equipment maintenance and requires specialized separation steps.
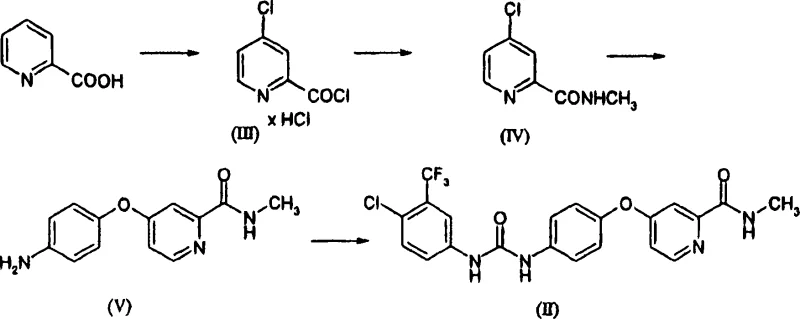
The Novel Approach
The process disclosed in CN101052619A fundamentally re-engineers the synthesis to overcome these bottlenecks. In the initial activation step, the invention eliminates the need for DMF by introducing a bromine compound catalyst, such as hydrogen bromide or sodium bromide, during the reaction with thionyl chloride. This modification not only prevents the formation of corrosive byproducts but also allows for precise control over gas evolution, enhancing safety profiles for large-scale reactors. Additionally, the urea coupling step is successfully migrated from dichloromethane to non-chlorinated solvents like ethyl acetate or tetrahydrofuran, aligning the process with modern green chemistry principles. Perhaps most critically, the final salt formation introduces a unique solvent engineering strategy where controlled amounts of water are added to improve solubility for filtration, a counter-intuitive yet highly effective technique for maximizing recovery rates without compromising crystal quality.
Mechanistic Insights into Urea Formation and Salt Crystallization
The formation of the urea linkage in Formula (II) involves the nucleophilic attack of the aniline nitrogen in Formula (V) onto the electrophilic carbon of the 4-chloro-3-trifluoromethylphenyl isocyanate. The patent specifies maintaining the reaction temperature below 70°C, preferably between 20°C and 60°C, to prevent side reactions such as the polymerization of the isocyanate or the formation of biuret impurities. The choice of a non-chlorinated solvent inert to isocyanates, such as ethyl acetate, is crucial because it provides the right balance of polarity to dissolve the reactants while facilitating the precipitation of the product upon cooling. This precipitation-driven isolation minimizes the need for chromatographic purification, which is often a bottleneck in kilogram-scale operations. The mechanistic efficiency here relies on the precise stoichiometric control of the isocyanate, typically used in a slight excess (1.0 to 2.0 equivalents) to drive the reaction to completion while ensuring that unreacted isocyanate can be quenched or removed easily during workup.
The subsequent conversion to the tosylate salt (Formula I) exhibits a fascinating solubility phenomenon that is key to the process's success. Typically, adding water to an organic solution of a hydrophobic drug substance would induce immediate precipitation, potentially trapping impurities. However, this patent reveals that adding a specific ratio of water (e.g., solvent-to-water ratios of 4:1 to 60:1) initially increases the solubility of the free base or intermediate species, allowing for a hot clarification filtration to remove insoluble particulates. Only after this clarification is the full amount of p-toluenesulfonic acid added, or the temperature lowered, to induce crystallization. This mechanism ensures that the final crystal lattice forms around a纯净 solution, significantly reducing the inclusion of mother liquor impurities and resulting in a product with purity levels exceeding 99%, which is critical for meeting the stringent specifications of high-purity pharmaceutical intermediates.
How to Synthesize Sorafenib Intermediate Efficiently
The synthesis outlined in the patent provides a clear roadmap for transitioning from laboratory benchtop procedures to commercial manufacturing. The process is designed to be telescoped where possible, such as using the crude acid chloride solution directly in the amidation step without isolation, thereby reducing cycle times and solvent consumption. The detailed protocol emphasizes the importance of temperature control during exothermic steps, particularly the addition of thionyl chloride and the subsequent urea coupling. For process chemists looking to implement this route, the key lies in replicating the specific crystallization parameters for the tosylate salt, including the seeding temperature and the rate of cooling, to ensure consistent particle size distribution and filterability. The following guide summarizes the critical operational phases derived from the patent examples.
- React 2-pyridinecarboxylic acid with thionyl chloride and a bromine catalyst to form the acid chloride intermediate without using DMF.
- Convert the acid chloride to the N-methyl amide using aqueous methylamine or gaseous methylamine in a water-immiscible solvent.
- Perform nucleophilic aromatic substitution with 4-aminophenol using a base, optionally isolating the dihydrochloride salt for purification.
- React the amino-ether intermediate with 4-chloro-3-trifluoromethylphenyl isocyanate in a non-chlorinated solvent like ethyl acetate.
- Crystallize the final tosylate salt by adding p-toluenesulfonic acid in a polar solvent, utilizing controlled water addition to enhance solubility and yield.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the technical improvements in CN101052619A translate directly into tangible business benefits, primarily through risk mitigation and operational efficiency. The shift away from dichloromethane and DMF reduces the regulatory burden associated with solvent residue limits and hazardous waste disposal, which are significant cost drivers in API manufacturing. By adopting a route that utilizes safer, more common solvents like ethyl acetate and toluene, manufacturers can secure more stable raw material supplies and avoid the price volatility often seen with regulated chlorinated solvents. Furthermore, the ability to perform clarification filtrations prior to crystallization reduces the likelihood of batch failures due to particulate contamination, ensuring a more reliable delivery schedule for downstream customers.
- Cost Reduction in Manufacturing: The elimination of DMF in the acid chloride step removes the need for complex distillation setups required to recover high-boiling solvents, leading to substantial energy savings. Additionally, the avoidance of extensive extraction steps in the etherification phase reduces solvent consumption and labor costs associated with phase separations. By simplifying the workup procedures and enabling the use of crude intermediates in subsequent steps, the overall processing time is drastically reduced, which lowers the fixed cost allocation per kilogram of product. These efficiencies compound to offer a significantly lower cost of goods sold (COGS) compared to legacy methods.
- Enhanced Supply Chain Reliability: The robustness of the new synthetic route enhances supply continuity by minimizing the number of unit operations where yield losses typically occur. The use of aqueous methylamine and the ability to handle exotherms safely with bromine catalysts make the process less susceptible to minor variations in raw material quality or reactor performance. This stability is crucial for maintaining long-term contracts with API manufacturers who require guaranteed volumes. Moreover, the simplified purification strategy reduces the dependency on specialized chromatography resins or adsorbents, which can sometimes face global supply shortages, thereby de-risking the entire production timeline.
- Scalability and Environmental Compliance: From an environmental perspective, the replacement of chlorinated solvents with esters and ethers aligns the manufacturing process with increasingly strict global environmental regulations. This proactive compliance future-proofs the supply chain against potential bans or taxes on volatile organic compounds (VOCs). The improved volumetric yield, achieved through the optimized crystallization protocol, means that existing reactor capacity can produce more product without the need for capital expenditure on new equipment. This scalability ensures that the supplier can rapidly ramp up production to meet surges in demand for oncology therapies without compromising on quality or safety standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. These insights are derived directly from the experimental data and claims within CN101052619A, providing clarity on how this method compares to standard industry practices. Understanding these details helps stakeholders make informed decisions about technology transfer and vendor qualification.
Q: How does the new process improve safety compared to conventional methods?
A: The novel process eliminates the use of dimethylformamide (DMF) in the acid chloride formation step by utilizing a bromine compound catalyst with thionyl chloride. This avoids the formation of corrosive dimethylcarbamoyl chloride and allows for better control of gas evolution (SO2 and HCl), significantly enhancing operational safety on an industrial scale.
Q: What is the key innovation in the final crystallization step?
A: The patent describes a counter-intuitive method where water is added to the polar solvent system during the reaction with p-toluenesulfonic acid. Although the compound has low water solubility, this specific addition increases the solubility of the intermediate enough to allow for clarification filtration before crystallization, leading to higher volumetric yield and purity.
Q: Can chlorinated solvents be avoided in the urea formation step?
A: Yes, unlike prior art methods that rely on dichloromethane (DCM), this invention successfully utilizes non-chlorinated organic solvents such as ethyl acetate or tetrahydrofuran for the reaction between the amine intermediate and the isocyanate. This reduces environmental impact and simplifies solvent recovery processes.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sorafenib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the complexity of synthesizing kinase inhibitor intermediates requires more than just standard chemical capability; it demands deep process understanding and rigorous quality control. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from pilot plant to full-scale manufacturing is seamless. We are committed to delivering products with stringent purity specifications, utilizing our rigorous QC labs to verify every batch against the highest international standards. Our infrastructure is designed to handle the specific solvent systems and safety requirements outlined in advanced patents like CN101052619A, guaranteeing a supply that is both consistent and compliant.
We invite potential partners to engage with our technical procurement team to discuss how our manufacturing capabilities can support your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain a clearer picture of how our optimized processes can reduce your overall procurement costs. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your supply chain requirements, ensuring that your development timelines are met with precision and reliability.