Revolutionizing Quinazolinone Production: A Green, Metal-Free Synthetic Route for Commercial Scale
Revolutionizing Quinazolinone Production: A Green, Metal-Free Synthetic Route for Commercial Scale
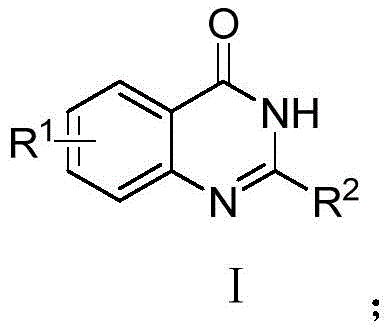
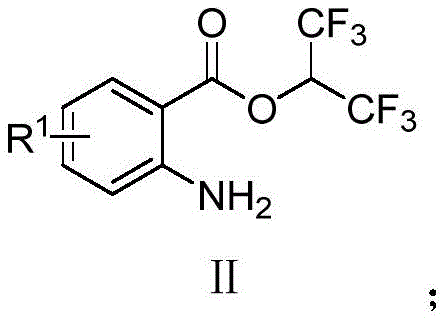
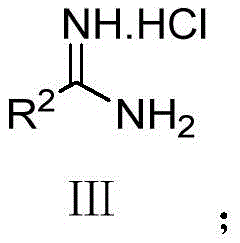
The pharmaceutical and fine chemical industries are constantly seeking methodologies that balance high efficiency with environmental sustainability, a challenge elegantly addressed by the technology disclosed in Chinese Patent CN112608281A. This patent introduces a groundbreaking green synthesis method for quinazolinone compounds, a privileged scaffold ubiquitous in medicinal chemistry due to its diverse biological activities ranging from antimalarial to anticancer properties. Unlike traditional synthetic routes that often rely on elevated temperatures and transition metal catalysts, this novel approach utilizes substituted 2-aminobenzoic acid hexafluoroisopropanol esters and amidine hydrochlorides as key building blocks. The reaction proceeds smoothly at room temperature in common polar aprotic solvents, achieving near-quantitative conversion rates without generating hazardous by-products. For R&D directors and process chemists, this represents a paradigm shift towards atom-economical and operationally simple heterocyclic construction, while supply chain managers will appreciate the elimination of expensive catalytic systems and energy-intensive heating steps.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinazolinone core has been fraught with synthetic inefficiencies that pose significant hurdles for large-scale manufacturing and cost-effective procurement. Conventional methods frequently necessitate the use of harsh reaction conditions, including prolonged heating at high temperatures and the employment of stoichiometric amounts of toxic transition metal catalysts or strong acidic promoters. These aggressive conditions not only increase energy consumption and operational costs but also complicate the downstream purification process, often requiring rigorous removal of trace metal residues to meet stringent pharmaceutical quality standards. Furthermore, many traditional pathways suffer from poor atom economy, generating substantial quantities of chemical waste and requiring complex work-up procedures such as column chromatography to isolate the pure product. Such limitations severely impact the scalability of the process and inflate the final cost of goods, making it difficult for procurement teams to secure reliable supplies of high-purity intermediates at competitive prices.
The Novel Approach
In stark contrast to these legacy methods, the invention detailed in CN112608281A offers a remarkably mild and efficient alternative that leverages the unique reactivity of hexafluoroisopropanol esters. By employing these activated esters as electrophiles, the reaction can be driven to completion at ambient temperature using simple inorganic bases like potassium phosphate, completely obviating the need for external heating or metal catalysis. This strategic choice of starting materials ensures that the reaction proceeds with exceptional selectivity, yielding the desired quinazolinone structure with minimal formation of side products or impurities. The simplicity of the post-reaction work-up, which involves basic extraction and washing rather than complex chromatographic separation, drastically reduces solvent usage and processing time. This streamlined protocol not only enhances the overall yield, often reaching up to 99%, but also aligns perfectly with the principles of green chemistry, offering a sustainable solution for the commercial production of complex heterocyclic intermediates.
Mechanistic Insights into Base-Promoted Cyclization
The mechanistic elegance of this synthesis lies in the dual nucleophilic character of the amidine species and the enhanced leaving group ability of the hexafluoroisopropoxy moiety. Upon addition of the base, the amidine hydrochloride is deprotonated to generate a free nucleophilic amidine, which subsequently attacks the carbonyl carbon of the 2-aminobenzoic acid hexafluoroisopropanol ester. This initial nucleophilic acyl substitution results in the expulsion of the hexafluoroisopropoxide anion, a highly stable leaving group due to the strong electron-withdrawing effect of the trifluoromethyl groups, forming an intermediate amide. Following this acylation, an intramolecular nucleophilic attack occurs where the ortho-amino group on the benzene ring attacks the imine carbon of the amidine moiety. This cyclization step, followed by the elimination of ammonia gas, closes the heterocyclic ring to form the stable quinazolinone core. The entire cascade is thermodynamically favorable and kinetically accessible at room temperature, ensuring high conversion efficiency without the need for thermal activation.
From an impurity control perspective, this mechanism offers distinct advantages over metal-catalyzed alternatives. Since no transition metals are involved, there is zero risk of metal contamination in the final API intermediate, a critical parameter for regulatory compliance in pharmaceutical manufacturing. Additionally, the mild conditions prevent the degradation of sensitive functional groups that might be present on the aromatic rings, such as halogens or nitro groups, thereby preserving the structural integrity of diverse derivatives. The use of hexafluoroisopropanol esters also minimizes the formation of polymeric by-products often seen in high-temperature condensations. Consequently, the crude reaction mixture is exceptionally clean, allowing for isolation of the product with high purity simply through aqueous work-up. This robustness makes the process highly suitable for generating libraries of analogs for SAR studies, as demonstrated by the successful synthesis of various substituted derivatives shown in the patent examples.
How to Synthesize Quinazolinone Derivatives Efficiently
The practical implementation of this green synthesis route is straightforward and requires standard laboratory equipment, making it easily transferable from bench scale to pilot plant operations. The process begins with the precise weighing of the substituted 2-aminobenzoic acid hexafluoroisopropanol ester and the corresponding amidine hydrochloride salt, typically in a molar ratio of 1:1.2 to ensure complete consumption of the ester. These solids are suspended in a polar aprotic solvent, with DMF identified as the optimal medium for maximizing yield, although acetonitrile and dioxane are also viable alternatives. The addition of a base, preferably potassium phosphate, initiates the reaction immediately at room temperature, and the mixture is stirred for approximately 8 to 12 hours to ensure full conversion. Detailed standardized operating procedures for scaling this reaction, including specific quenching and isolation protocols, are outlined in the technical guide below.
- Prepare the reaction mixture by combining R1-substituted 2-aminobenzoic acid hexafluoroisopropanol ester and R2-substituted amidine hydrochloride in a polar aprotic solvent like DMF.
- Add an inorganic or organic base, preferably potassium phosphate (K3PO4), to the mixture to facilitate the deprotonation and nucleophilic attack.
- Stir the reaction at room temperature for 8-12 hours, followed by extraction with ethyl acetate, water washing, and drying to isolate the high-purity quinazolinone product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic methodology translates into tangible strategic benefits that extend far beyond simple yield improvements. The elimination of transition metal catalysts removes a significant cost center associated with both the purchase of expensive metals like palladium or copper and the subsequent validation of their removal from the final product. This simplification of the supply chain reduces dependency on volatile metal markets and mitigates the risk of supply disruptions caused by catalyst shortages. Furthermore, the ability to run the reaction at room temperature significantly lowers the energy footprint of the manufacturing process, contributing to reduced utility costs and a smaller carbon footprint, which is increasingly important for meeting corporate sustainability goals. The simplified work-up procedure also means faster batch turnover times, allowing manufacturing facilities to increase throughput without requiring additional capital investment in new reactor hardware.
- Cost Reduction in Manufacturing: The economic impact of this process is profound, primarily driven by the removal of costly catalytic systems and the reduction in solvent consumption during purification. By avoiding the need for column chromatography and relying instead on simple extraction and crystallization, the volume of organic solvents required per kilogram of product is drastically reduced, leading to lower waste disposal costs and reduced raw material expenditure. Additionally, the high atom economy of the reaction ensures that the majority of the starting material mass is incorporated into the final product, minimizing waste generation. These factors combined result in a significantly lower cost of goods sold (COGS), providing a competitive pricing advantage in the global market for pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The robustness of this room-temperature protocol enhances supply chain resilience by reducing the complexity of the manufacturing process. Since the reaction does not require specialized heating equipment or strict inert atmosphere conditions often needed for air-sensitive metal catalysts, it can be performed in a wider range of manufacturing facilities, increasing the pool of potential contract manufacturing organizations (CMOs). The stability of the hexafluoroisopropanol ester starting materials also ensures consistent quality and availability, reducing the risk of batch-to-batch variability that can plague more sensitive synthetic routes. This reliability is crucial for maintaining continuous production schedules and meeting tight delivery deadlines for downstream API manufacturers.
- Scalability and Environmental Compliance: Scaling this green synthesis from laboratory to industrial production is seamless due to the absence of exothermic hazards associated with rapid heating or highly reactive reagents. The mild reaction conditions allow for safe operation in large-scale reactors without the need for complex cooling systems to manage heat release. Moreover, the process aligns with stringent environmental regulations by minimizing the generation of hazardous waste and avoiding the use of toxic heavy metals. This environmental compliance simplifies the permitting process for new manufacturing lines and reduces the long-term liability associated with waste management, making it an attractive option for companies committed to sustainable chemical manufacturing practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this quinazolinone synthesis technology. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the practical aspects of the method. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their existing production workflows or for procurement specialists assessing the quality and consistency of the supplied intermediates.
Q: What are the primary advantages of using hexafluoroisopropanol esters in quinazolinone synthesis?
A: The use of hexafluoroisopropanol esters acts as a superior leaving group compared to traditional halides or carboxylic acids, enabling the reaction to proceed at room temperature without the need for harsh heating or toxic metal catalysts, thereby simplifying purification.
Q: Does this green synthesis method require column chromatography for purification?
A: No, one of the key commercial benefits of this patented process is that the crude product can be purified simply through extraction and water washing, eliminating the need for time-consuming and solvent-intensive column chromatography.
Q: What is the biological activity profile of the synthesized quinazolinone derivatives?
A: The synthesized compounds exhibit potent antitumor activity, specifically showing strong inhibition against breast cancer cells (MCF7), with certain derivatives demonstrating efficacy superior to the standard drug 5-Fluorouracil.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the green synthesis methods described in CN112608281A and have integrated similar advanced technologies into our own R&D and production capabilities. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive consistent, high-quality intermediates regardless of volume. Our commitment to excellence is underpinned by stringent purity specifications and rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify the identity and purity of every batch. We understand that in the pharmaceutical industry, the reliability of your supply chain is paramount, and we strive to be the dependable partner that supports your drug development journey from early-stage discovery through to commercial launch.
We invite you to collaborate with us to leverage these innovative synthetic routes for your specific project needs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your target molecule, demonstrating how our optimized processes can reduce your overall development costs. Please contact our technical procurement team today to request specific COA data for our quinazolinone portfolio or to discuss route feasibility assessments for your custom synthesis projects. Let us help you accelerate your timeline to market with efficient, scalable, and compliant chemical solutions.