Advanced Purification Technology for Lovastatin and Simvastatin Commercial Scale-up
The pharmaceutical industry continuously seeks robust methodologies to ensure the highest purity standards for critical cholesterol-lowering agents. Patent CN1443182A introduces a transformative approach for purifying lovastatin and simvastatin, specifically targeting the reduction of dimeric impurities to levels below 0.08%. This technical breakthrough addresses a persistent challenge in statin manufacturing where traditional lactonization processes often yield unwanted oligomeric byproducts that compromise drug safety and efficacy. By employing a selective hydrolysis strategy using mild bases, manufacturers can achieve superior impurity profiles without compromising the integrity of the lactone ring. This innovation is particularly relevant for partners seeking a reliable pharmaceutical intermediates supplier who can deliver high-purity materials consistent with stringent global regulatory requirements. The structural complexity of these molecules demands precise control, as illustrated by the chemical architecture of the target compounds.
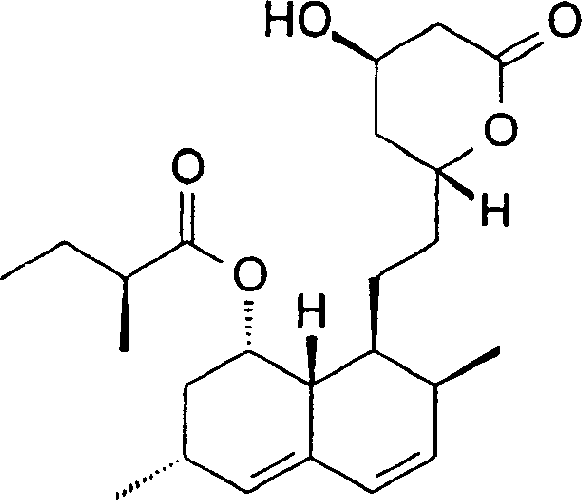
Furthermore, the applicability of this purification protocol extends to simvastatin, a synthetic analog with enhanced potency, ensuring that both key molecules in the statin class benefit from this refined processing technique. The ability to consistently produce materials with such low impurity thresholds significantly de-risks the downstream formulation process for drug developers. For R&D teams, this means a more predictable impurity spectrum, facilitating smoother regulatory filings and reducing the burden on analytical quality control departments. The strategic implementation of this patent-protected method represents a significant leap forward in process chemistry, aligning perfectly with the needs of modern pharmaceutical supply chains that prioritize quality and consistency above all else.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the production of statin lactones has relied heavily on lactonization processes that involve harsh conditions, such as high-temperature reflux in inert solvents or the use of strong mineral acids at ambient temperatures. These conventional methods are inherently problematic because the lactonization reaction is an equilibrium process that competes with intermolecular esterification, leading to the formation of dimeric and oligomeric impurities. Once formed, these dimers are notoriously difficult to remove because they co-crystallize with the desired product, often resulting in final purity levels that hover around 95-98% with dimer content ranging from 0.08% to 0.4%. To mitigate this, manufacturers have previously resorted to high-dilution techniques, which drastically reduce process efficiency and increase solvent consumption, making industrial scale-up economically and environmentally unsustainable. Additionally, the use of strong corrosive acids necessitates complex neutralization steps and generates significant chemical waste, creating bottlenecks in production throughput and increasing the overall environmental footprint of the manufacturing facility.
The Novel Approach
In stark contrast, the novel approach detailed in the patent data utilizes a mild base treatment in a carefully selected solvent mixture to selectively hydrolyze these stubborn dimeric impurities. This method operates under significantly milder conditions, avoiding the extreme thermal stress and corrosive environments associated with traditional techniques. By leveraging the differential reactivity between the dimeric esters and the monomeric lactone ring, the process effectively cleaves the impurities into water-soluble hydroxy acids while leaving the active pharmaceutical ingredient intact. This selectivity is the cornerstone of the innovation, allowing for a dramatic reduction in impurity levels to below 0.08% without the need for excessive dilution or hazardous reagents. The result is a streamlined workflow that not only enhances product quality but also simplifies the overall manufacturing process, offering a clear pathway for cost reduction in pharmaceutical manufacturing through improved yield and reduced waste disposal requirements.
Mechanistic Insights into Selective Base-Catalyzed Hydrolysis
The core mechanism driving this purification success lies in the nuanced chemical behavior of esters under mildly alkaline conditions. The dimeric impurities, formed via intermolecular esterification during prior synthesis steps, possess ester linkages that are susceptible to hydrolysis by mild bases such as ammonium hydroxide or aliphatic amines. Crucially, the reaction conditions are tuned so that the base strength and exposure time are sufficient to cleave the dimer esters but insufficient to open the intramolecular lactone ring of the lovastatin or simvastatin molecule. This kinetic selectivity ensures that the active drug substance remains stable while the impurities are converted into their corresponding hydroxy acid forms, which exhibit vastly different solubility profiles in the chosen solvent system. The reaction equilibrium is thus shifted favorably towards the purification of the lactone, as the hydrolyzed impurities remain in the mother liquor during the subsequent crystallization step. Understanding this mechanistic distinction is vital for process chemists aiming to replicate these results, as it highlights the importance of precise base equivalence and temperature control.
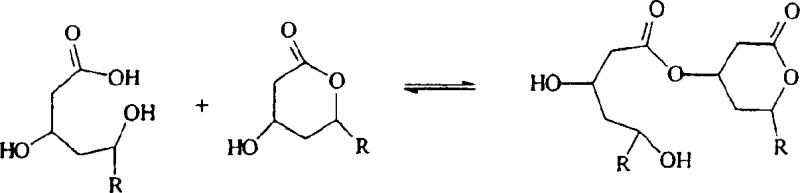
Furthermore, the choice of solvent mixture plays a pivotal role in stabilizing the lactone form against unwanted hydrolysis while facilitating the dissolution of the crude material. Solvents like isobutyl acetate mixed with ethanol create an environment where the lactone product has limited solubility upon cooling, promoting high-yield crystallization, while the hydrolyzed impurities remain soluble. This differential solubility is the physical manifestation of the chemical selectivity achieved in the reaction phase. The process effectively decouples the purification from the synthesis, allowing manufacturers to treat crude batches that may have higher initial impurity loads and still achieve specification-grade purity. For technical teams, this means greater flexibility in upstream fermentation or synthesis parameters, as the downstream purification step is robust enough to handle variability. The mechanistic elegance of this approach ensures that the final product meets the rigorous demands of the global market for high-purity pharmaceutical intermediates.
How to Synthesize Lovastatin Efficiently
Implementing this purification strategy requires a disciplined approach to solvent selection and temperature management to maximize the selective hydrolysis effect. The process begins by dissolving the crude statin lactone in a specific ratio of ester and alcohol solvents, followed by the controlled addition of a mild base reagent. Maintaining the reaction temperature within the optimal range is critical to ensure that the hydrolysis of dimers proceeds to completion without triggering the degradation of the main product. Once the reaction is complete, a controlled cooling profile is applied to induce crystallization of the purified lactone, leaving the impurities in the solution. The detailed standardized synthesis steps see the guide below for specific operational parameters and safety considerations.
- Dissolve crude lovastatin or simvastatin lactone in a solvent mixture such as isobutyl acetate and ethanol.
- Add a mild base reagent like ammonium hydroxide to the solution at controlled temperatures between 40-85°C.
- Cool the mixture gradually to crystallize the purified lactone while leaving hydrolyzed dimer impurities in the solution.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this purification technology offers substantial benefits that resonate deeply with procurement and supply chain leadership. The elimination of harsh reagents and the reduction of complex neutralization steps translate directly into a safer working environment and lower operational overheads. By simplifying the purification workflow, manufacturers can achieve faster batch turnover times, which enhances the overall responsiveness of the supply chain to market demands. The ability to consistently produce material with ultra-low impurity levels reduces the risk of batch rejection, thereby securing supply continuity for downstream drug manufacturers. This reliability is a key differentiator in the competitive landscape of API intermediates, where consistency is often valued higher than marginal price differences. Furthermore, the reduced environmental burden aligns with increasingly strict global sustainability mandates, future-proofing the supply chain against regulatory tightening.
- Cost Reduction in Manufacturing: The process eliminates the need for expensive strong acid catalysts and the associated waste treatment costs required to neutralize them. By avoiding high-dilution techniques, solvent consumption is significantly reduced, leading to lower raw material expenses and reduced costs for solvent recovery or disposal. The higher yield obtained through improved crystallization efficiency means more saleable product per batch, effectively lowering the cost of goods sold. Additionally, the simplified workflow reduces labor hours and equipment usage time, contributing to overall operational efficiency. These qualitative improvements collectively drive a more competitive cost structure without compromising on the quality standards required for pharmaceutical applications.
- Enhanced Supply Chain Reliability: The robustness of the mild base purification method ensures that production schedules are less likely to be disrupted by complex troubleshooting or re-processing of off-spec batches. The use of common, readily available solvents and reagents minimizes the risk of supply bottlenecks for raw materials. This stability allows for more accurate forecasting and inventory management, ensuring that customers receive their orders on time. The consistent quality output reduces the need for extensive incoming quality testing by customers, streamlining the intake process. Ultimately, this reliability builds stronger long-term partnerships between suppliers and pharmaceutical companies, fostering a stable and predictable supply environment.
- Scalability and Environmental Compliance: The mild reaction conditions make this process inherently safer and easier to scale from pilot plant to full commercial production without significant engineering hurdles. The reduction in hazardous waste generation simplifies compliance with environmental regulations, reducing the administrative and financial burden of waste management. The process design supports green chemistry principles by minimizing energy consumption through lower temperature requirements and reducing the use of toxic substances. This alignment with environmental best practices enhances the corporate social responsibility profile of the manufacturing operation. Scalability is further supported by the use of standard crystallization equipment, ensuring that capacity can be expanded to meet growing market demand for high-purity statins.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this purification technology. These insights are derived directly from the patent specifications and are intended to clarify the operational benefits and chemical principles involved. Understanding these details helps stakeholders make informed decisions about integrating this method into their existing manufacturing frameworks. The answers reflect the consensus on best practices for achieving optimal purity and yield while maintaining safety and compliance standards.
Q: How does this process reduce dimeric impurities in statins?
A: The process utilizes a mild base to selectively hydrolyze dimeric impurities into water-soluble forms without opening the lactone ring of the active pharmaceutical ingredient.
Q: What are the advantages over conventional strong acid lactonization?
A: Unlike strong acid methods that require neutralization and generate waste, this mild base approach operates under safer conditions and simplifies downstream processing.
Q: Can this method be scaled for industrial production?
A: Yes, the use of common solvents and mild reagents makes the process highly suitable for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Lovastatin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of purity and process reliability in the production of life-saving medications. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated purification methods described in patent CN1443182A can be seamlessly integrated into our manufacturing lines. We are committed to maintaining stringent purity specifications and operating rigorous QC labs to verify that every batch meets the < 0.08% dimer impurity threshold. Our infrastructure is designed to handle complex chemical transformations with precision, providing our partners with a secure source of high-quality pharmaceutical intermediates. We understand that consistency is the backbone of a successful drug supply chain, and we invest heavily in process optimization to deliver exactly that.
We invite you to engage with our technical procurement team to discuss how our capabilities align with your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits of switching to our purified materials. We encourage potential partners to contact us for specific COA data and route feasibility assessments to validate our performance against your internal standards. Let us collaborate to optimize your supply chain and ensure the uninterrupted availability of high-purity statin intermediates for your global operations.