Advanced Synthesis of Alpha-Substituted Tetrahydro-Gamma-Carboline Compounds for Oncology Drug Development
Introduction to Next-Generation Carboline Synthesis
The pharmaceutical industry is constantly seeking efficient pathways to access novel heterocyclic scaffolds that possess potent biological activity. A significant breakthrough in this domain is detailed in Chinese Patent CN110698474B, which discloses a robust and versatile method for synthesizing alpha-substituted tetrahydro-gamma-carboline compounds. Unlike their beta-carboline counterparts which have been extensively studied, gamma-carbolines represent an underexplored chemical space with immense potential for drug discovery, particularly in oncology. This patent introduces a streamlined synthetic strategy that bypasses the limitations of traditional multi-step syntheses, enabling the rapid generation of diverse molecular libraries.
The core innovation lies in the direct functionalization of the alpha-position of the tetrahydro-gamma-carboline skeleton. This transformation is achieved through a mild electrophilic activation followed by nucleophilic substitution, a process that maintains the integrity of sensitive functional groups while delivering high yields. The resulting compounds have demonstrated significant inhibitory activity against MCF-7 and A549 cancer cell lines, validating their utility as high-purity pharmaceutical intermediates. For research and development teams, this methodology offers a powerful tool to accelerate lead optimization programs without the burden of complex process chemistry.
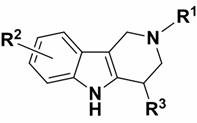
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the functionalization of carboline scaffolds at specific positions has been a formidable challenge for process chemists. Conventional routes often rely on harsh reaction conditions, such as strong acidic or basic environments, elevated temperatures, and high-pressure systems, which can lead to decomposition of the sensitive indole moiety or unwanted side reactions. Furthermore, traditional strategies frequently necessitate the use of protecting groups to mask reactive sites, adding multiple steps to the synthesis sequence. This not only increases the overall cost of goods but also reduces the overall atom economy and generates substantial chemical waste. The requirement for transition metal catalysts in some cross-coupling approaches introduces additional complexities regarding heavy metal removal and regulatory compliance for final drug substances.
The Novel Approach
The methodology described in the patent represents a paradigm shift towards green and efficient synthesis. By utilizing tert-butyl hypochlorite (t-BuOCl) as a mild activating agent, the process generates a reactive intermediate at the alpha-position under ambient conditions. This activated species is then immediately trapped by a wide variety of nucleophiles in a one-pot fashion. The reaction proceeds rapidly at room temperature, typically within minutes to a few hours, eliminating the need for energy-intensive heating or cooling cycles. This approach drastically simplifies the operational workflow, as it avoids the use of expensive catalysts and complex purification protocols associated with metal residues. The versatility of this method is exemplified by its compatibility with diverse nucleophiles, ranging from simple anilines to complex heterocycles.
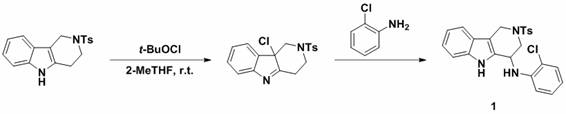
Mechanistic Insights into t-BuOCl Mediated Alpha-Functionalization
The chemical mechanism underpinning this transformation involves the generation of an electrophilic species at the alpha-carbon of the tetrahydro-gamma-carboline ring system. Upon addition of tert-butyl hypochlorite to the solution of the starting material in a solvent like 2-methyltetrahydrofuran (2-MeTHF), an initial chlorination or iminium ion formation likely occurs. This activation step renders the alpha-position highly susceptible to nucleophilic attack. The subsequent addition of the nucleophile, whether it be a nitrogen-based amine or a carbon-based indole, leads to the displacement of the leaving group or addition across the activated double bond, restoring aromaticity or forming the stable substituted product. The mildness of t-BuOCl ensures that over-oxidation or degradation of the indole core is minimized, preserving the structural fidelity required for biological activity.
From an impurity control perspective, this mechanism offers distinct advantages. The reaction is highly selective for the alpha-position due to the electronic properties of the carboline nitrogen and the adjacent carbon atoms. This regioselectivity minimizes the formation of positional isomers, which are often difficult to separate and can complicate the regulatory filing process. Furthermore, the use of 2-MeTHF, a bio-based solvent with favorable extraction properties, facilitates easy workup and reduces the risk of solvent-related impurities. The broad substrate scope allows for the introduction of various functional groups, such as halogens, trifluoromethyl groups, and nitro groups, without compromising the reaction efficiency, thereby enabling the fine-tuning of physicochemical properties for drug candidates.
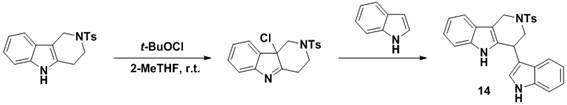
How to Synthesize Alpha-Substituted Tetrahydro-Gamma-Carboline Efficiently
Implementing this synthesis route in a laboratory or pilot plant setting requires attention to reagent quality and reaction monitoring to ensure optimal outcomes. The process begins with the dissolution of the tetrahydro-gamma-carboline precursor in an appropriate solvent, followed by the precise addition of the activating agent. The short reaction time necessitates careful monitoring, typically via thin-layer chromatography (TLC), to prevent over-reaction. Once the nucleophile is added, the mixture is stirred until conversion is complete, after which standard aqueous workup and chromatographic purification yield the target compound. The detailed standardized synthesis steps are provided in the guide below.
- Dissolve the tetrahydro-gamma-carboline starting material in 2-methyltetrahydrofuran (2-MeTHF) or dichloromethane at room temperature.
- Add tert-butyl hypochlorite (t-BuOCl) to the solution and stir for approximately 2 minutes to activate the alpha position.
- Introduce the nucleophilic reagent (such as aniline, indole, or amine derivatives), monitor reaction completion via TLC, and purify the target product using silica gel chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers compelling economic and logistical benefits. The reliance on commodity chemicals like tert-butyl hypochlorite and 2-methyltetrahydrofuran ensures a stable and cost-effective supply of raw materials, mitigating the risks associated with sourcing exotic or proprietary reagents. The elimination of transition metal catalysts removes the need for expensive scavenging resins and extensive analytical testing for heavy metals, directly contributing to cost reduction in API manufacturing. Additionally, the ambient temperature operation significantly lowers energy consumption compared to processes requiring reflux or cryogenic conditions, aligning with corporate sustainability goals and reducing utility overheads.
- Cost Reduction in Manufacturing: The streamlined one-pot nature of this reaction eliminates multiple isolation and purification steps that are typical in traditional multi-step syntheses. By reducing the number of unit operations, manufacturers can achieve substantial cost savings in labor, solvent usage, and equipment time. The high atom economy and excellent yields reported in the patent examples further enhance the overall process efficiency, ensuring that more of the starting material is converted into valuable product rather than waste. This efficiency translates directly into a lower cost per kilogram for the final pharmaceutical intermediate.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions means that the process is less sensitive to minor fluctuations in temperature or reagent quality, leading to consistent batch-to-batch reproducibility. This reliability is crucial for maintaining continuous supply chains and meeting strict delivery timelines for downstream drug development projects. The use of widely available solvents and reagents also reduces the risk of supply disruptions caused by geopolitical issues or single-source dependencies, ensuring a secure supply of high-purity tetrahydro-gamma-carboline derivatives for global markets.
- Scalability and Environmental Compliance: Scaling this process from gram to kilogram or ton scale is straightforward due to the absence of hazardous high-pressure or high-temperature steps. The exothermic nature of the activation step is manageable with standard cooling protocols, making it safe for large-scale production. Furthermore, the choice of 2-MeTHF as a solvent supports environmental compliance initiatives, as it is derived from renewable resources and has a favorable safety profile compared to chlorinated solvents. This alignment with green chemistry principles simplifies waste disposal and regulatory reporting, facilitating faster approval for commercial scale-up of complex heterocyclic compounds.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these specialized carboline intermediates. The answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for industry professionals. Understanding these details is essential for evaluating the feasibility of integrating this technology into existing drug discovery pipelines.
Q: What is the primary advantage of this synthesis method over conventional routes?
A: The primary advantage is the exceptionally mild reaction conditions. Unlike traditional methods that often require harsh acids, high temperatures, or complex multi-step protection strategies, this patent-disclosed route operates at room temperature using common reagents like tert-butyl hypochlorite, significantly simplifying the operational workflow and reducing energy consumption.
Q: What types of nucleophiles are compatible with this alpha-functionalization strategy?
A: The method demonstrates remarkable substrate universality. It successfully accommodates a wide range of nucleophiles including various substituted anilines (electron-withdrawing and electron-donating groups), benzylamines, alkylamines, heterocyclic amines like aminobenzothiophene dioxide, and even carbon nucleophiles such as indole derivatives, allowing for the rapid construction of diverse compound libraries.
Q: What are the biological applications of these alpha-substituted compounds?
A: These compounds exhibit potent antitumor activity, specifically showing inhibition against human breast cancer (MCF-7) and lung cancer (A549) cell lines. The structural modification at the alpha position enhances biological potency compared to the parent tetrahydro-gamma-carboline skeleton, making them valuable candidates for developing new oncology therapeutics.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alpha-Substituted Tetrahydro-Gamma-Carboline Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of accessing high-quality intermediates for the development of next-generation oncology therapeutics. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and speed. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of alpha-substituted tetrahydro-gamma-carboline meets the highest international standards for pharmaceutical applications.
We invite you to collaborate with our technical team to explore how this innovative synthesis route can accelerate your drug development timeline. By leveraging our expertise in process optimization and scale-up, we can provide a Customized Cost-Saving Analysis tailored to your specific project requirements. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us help you bring your life-saving medicines to market faster and more efficiently.