Advanced Synthesis of Alpha-Substituted Tetrahydro-Gamma-Carboline Intermediates for Oncology Drug Development
The pharmaceutical industry is constantly seeking robust and efficient pathways to access novel heterocyclic scaffolds that possess potent biological activities, particularly in the realm of oncology. Patent CN110698474A introduces a groundbreaking methodology for the preparation of alpha-substituted tetrahydro-gamma-carboline compounds, a class of alkaloids that has historically been less explored compared to their beta-counterparts despite their significant therapeutic potential. This innovation addresses the critical need for concise synthetic routes that can construct focused compound libraries with high structural diversity and purity. By leveraging a mild electrophilic chlorination strategy followed by nucleophilic substitution, the disclosed technology enables the late-stage functionalization of the tetrahydro-gamma-carboline core with exceptional atom economy. For research directors and procurement specialists alike, this patent represents a pivotal advancement in accessing high-purity pharmaceutical intermediates that are essential for developing next-generation anticancer agents targeting breast, lung, and prostate cancers.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic approaches to functionalized carboline derivatives often suffer from significant drawbacks that hinder their utility in modern drug discovery and process chemistry. Conventional methods frequently rely on harsh reaction conditions, including elevated temperatures, strong acidic or basic environments, and the use of expensive transition metal catalysts that require complex removal steps to meet stringent purity specifications. These rigorous conditions not only increase the operational costs and energy consumption but also limit the substrate scope, making it difficult to introduce sensitive functional groups without extensive protection and deprotection strategies. Furthermore, multi-step syntheses typical of older methodologies result in lower overall yields and generate substantial chemical waste, posing challenges for environmental compliance and supply chain sustainability. The inability to perform direct late-stage modifications on the carboline scaffold often forces chemists to restart syntheses from early intermediates, drastically extending lead times for new candidate molecules.
The Novel Approach
In stark contrast to these legacy techniques, the method described in patent CN110698474A offers a remarkably streamlined and versatile solution for constructing alpha-substituted tetrahydro-gamma-carbolines. The core innovation lies in the use of tert-butyl hypochlorite (t-BuOCl) as a mild chlorinating agent that activates the alpha-position of the tetrahydro-gamma-carboline ring system at room temperature within mere minutes. This activation step generates a highly reactive intermediate that readily undergoes nucleophilic substitution with a wide array of amines, including anilines, benzylamines, and even indole derivatives, without the need for external heating or pressure. 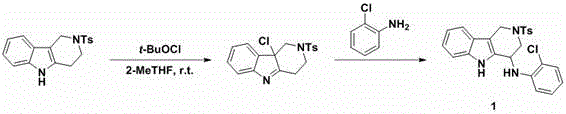 The operational simplicity of this protocol allows for rapid reaction monitoring via TLC and facilitates easy workup procedures, typically involving simple concentration and silica gel chromatography. This novel approach not only drastically reduces the reaction time from days to hours but also expands the chemical space accessible to medicinal chemists, enabling the rapid generation of diverse analogues for structure-activity relationship (SAR) studies with high efficiency.
The operational simplicity of this protocol allows for rapid reaction monitoring via TLC and facilitates easy workup procedures, typically involving simple concentration and silica gel chromatography. This novel approach not only drastically reduces the reaction time from days to hours but also expands the chemical space accessible to medicinal chemists, enabling the rapid generation of diverse analogues for structure-activity relationship (SAR) studies with high efficiency.
Mechanistic Insights into t-BuOCl-Mediated Alpha-Functionalization
The mechanistic pathway underpinning this transformation involves a sophisticated yet efficient sequence of electrophilic activation and nucleophilic attack that ensures high regioselectivity and yield. Initially, the tert-butyl hypochlorite acts as an electrophilic chlorine source, selectively targeting the electron-rich alpha-position adjacent to the nitrogen atom in the tetrahydro-gamma-carboline framework. This step proceeds rapidly at ambient temperature to form a transient alpha-chloro intermediate, which is stabilized by the adjacent nitrogen lone pair, preventing over-chlorination or degradation of the sensitive indole moiety. The mildness of this chlorination step is crucial, as it preserves other functional groups on the aromatic rings that might be susceptible to oxidation or hydrolysis under more aggressive conditions. Once formed, this activated species serves as an excellent electrophile for the subsequent substitution reaction.
Upon the addition of the nucleophile, such as a substituted aniline or an indole, the reaction proceeds through a concerted or stepwise substitution mechanism where the chloride leaving group is displaced by the nitrogen nucleophile. The presence of the electron-withdrawing or donating groups on the nucleophile can be finely tuned to modulate the reaction rate, yet the protocol demonstrates remarkable tolerance to various electronic environments, as evidenced by the successful synthesis of compounds with nitro, trifluoromethyl, and halogen substituents. This mechanistic robustness minimizes the formation of side products and impurities, which is a critical factor for maintaining high product purity without the need for recrystallization or preparative HPLC in many cases. The ability to control the reaction kinetics simply by adjusting the stirring time after t-BuOCl addition provides process chemists with a valuable handle for optimizing the impurity profile during scale-up operations.
How to Synthesize Alpha-Substituted Tetrahydro-Gamma-Carboline Efficiently
Implementing this synthesis route in a laboratory or pilot plant setting requires adherence to specific procedural details to maximize yield and safety while ensuring reproducibility across different batches. The process begins with the dissolution of the tetrahydro-gamma-carboline starting material in a green solvent such as 2-methyltetrahydrofuran (2-MeTHF) or dichloromethane, creating a homogeneous solution ready for activation. Precise stoichiometry of the tert-butyl hypochlorite is essential to drive the formation of the chlorinated intermediate without excess reagent that could complicate downstream purification. Following the brief activation period, the nucleophile is introduced, and the mixture is stirred until thin-layer chromatography confirms the complete consumption of the starting material.
- Dissolve the tetrahydro-gamma-carboline starting material in 2-methyltetrahydrofuran (2-MeTHF) or dichloromethane at room temperature.
- Add tert-butyl hypochlorite (t-BuOCl) to the solution and stir for 2 minutes to generate the reactive chlorinated intermediate.
- Introduce the chosen nucleophile (such as aniline or indole derivatives) and monitor the reaction progress via TLC until completion.
- Concentrate the reaction mixture under reduced pressure and purify the crude product using silica gel chromatography to obtain the target compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic methodology offers profound advantages for procurement managers and supply chain heads looking to optimize the cost structure and reliability of their intermediate sourcing. The elimination of expensive transition metal catalysts and the reliance on commodity chemicals like t-BuOCl and common solvents significantly lowers the raw material costs associated with production. Moreover, the room temperature operation eliminates the need for energy-intensive heating or cooling infrastructure, resulting in substantial utility savings and a reduced carbon footprint for the manufacturing process. The high atom economy and minimal waste generation align perfectly with modern green chemistry principles, simplifying waste disposal compliance and reducing environmental liabilities for the supplier.
- Cost Reduction in Manufacturing: The simplified workflow removes the need for complex protection group chemistry and expensive catalytic systems, directly translating to lower production costs per kilogram. By avoiding high-pressure reactors and specialized equipment, capital expenditure for manufacturing facilities is minimized, allowing for more competitive pricing structures for the final intermediates. The high yields reported in the patent examples, often exceeding 80% and reaching up to 91% for specific derivatives, ensure that raw material utilization is maximized, further driving down the cost of goods sold. This economic efficiency makes the technology highly attractive for large-scale commercial production where margin optimization is critical.
- Enhanced Supply Chain Reliability: The use of widely available and stable reagents ensures that the supply chain is resilient against disruptions caused by the scarcity of exotic chemicals. Since the reaction conditions are mild and forgiving, the risk of batch failure due to minor process deviations is significantly reduced, leading to more consistent delivery schedules for downstream customers. The versatility of the method allows for the production of a wide range of analogues from a common intermediate, enabling suppliers to respond quickly to changing demands in drug discovery pipelines without retooling entire production lines. This flexibility enhances the overall reliability of the supply chain for pharmaceutical partners.
- Scalability and Environmental Compliance: The protocol's compatibility with green solvents like 2-MeTHF supports sustainable manufacturing practices and eases regulatory approval processes in environmentally conscious markets. The absence of heavy metal residues simplifies the purification process and ensures that the final product meets stringent quality standards for pharmaceutical applications without additional cleaning steps. Scalability is inherently supported by the exothermic nature of the reaction being manageable at room temperature, allowing for safe expansion from gram to ton scales. This combination of safety, sustainability, and scalability positions the technology as a preferred choice for long-term commercial partnerships.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these novel carboline derivatives, providing clarity for potential partners and collaborators. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation to ensure accuracy and relevance. Understanding these details is crucial for evaluating the feasibility of integrating this technology into existing drug development programs.
Q: What are the typical reaction yields for this synthesis method?
A: According to patent CN110698474A, the reaction yields are generally high, often exceeding 60%, with specific examples demonstrating yields up to 91% under optimized conditions.
Q: Is this method suitable for large-scale production?
A: Yes, the process utilizes mild room temperature conditions and common solvents like 2-MeTHF, which facilitates safe scale-up and reduces energy consumption compared to high-temperature protocols.
Q: What biological activities do these compounds exhibit?
A: The synthesized alpha-substituted tetrahydro-gamma-carboline compounds have demonstrated significant inhibitory activity against MCF-7 breast cancer and A549 lung cancer cell lines.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alpha-Substituted Tetrahydro-Gamma-Carboline Supplier
As a leader in the fine chemical and pharmaceutical intermediate sector, NINGBO INNO PHARMCHEM is uniquely positioned to leverage this advanced synthetic technology to deliver high-quality alpha-substituted tetrahydro-gamma-carboline compounds to the global market. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the volume requirements of both clinical trial materials and commercial drug manufacturing. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instruments to guarantee that every batch meets the highest industry standards. Our commitment to quality and consistency makes us the ideal partner for pharmaceutical companies seeking reliable sources of complex oncology intermediates.
We invite you to contact our technical procurement team to discuss your specific project requirements and explore how our capabilities can accelerate your drug development timeline. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our optimized processes can reduce your overall project costs while maintaining superior quality. We are ready to provide specific COA data and route feasibility assessments to support your decision-making process and help you bring life-saving therapies to patients faster. Partner with us to secure a stable and efficient supply chain for your next-generation anticancer candidates.