Scalable Purification Technology for High-Purity Ezetimibe Intermediates
Scalable Purification Technology for High-Purity Ezetimibe Intermediates
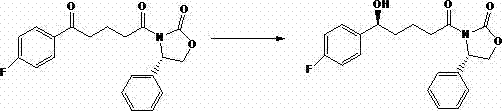
The pharmaceutical industry constantly seeks robust methodologies to ensure the highest quality of active pharmaceutical ingredients and their precursors. Patent CN102746249B introduces a groundbreaking purification refining method specifically designed for the intermediate of Ezetimibe, a critical cholesterol absorption inhibitor. This technology addresses the persistent challenges associated with removing trace impurities and residual reducing agents that often plague conventional synthesis routes. By leveraging a sophisticated solvent exchange strategy, the process achieves exceptional purity levels exceeding 99 percent and maintains an enantiomeric excess value greater than 99 percent. For global procurement leaders and R&D directors, this represents a significant leap forward in securing a reliable supply chain for high-value cardiovascular therapeutics. The method transforms what was once a difficult oily residue into a highly crystalline, pharmaceutically acceptable solid through precise control of solvent polarity and temperature.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional purification techniques for Ezetimibe intermediates often struggle with the complex physicochemical properties of the crude reaction mixture. In many prior art methods, the product obtained after the reduction step exists as an oily substance that is notoriously difficult to crystallize directly. This physical state traps significant amounts of impurities and residual reagents within the matrix, leading to products that fail to meet stringent quality standards required for human consumption. Furthermore, conventional recrystallization attempts frequently fail because the impurities possess solubility profiles very similar to the target molecule, making separation inefficient. The presence of residual chiral reducing agents and their decomposition products further complicates the purification landscape, often necessitating expensive and time-consuming chromatographic interventions. These limitations result in lower overall yields, increased production costs, and extended lead times that strain supply chain reliability for downstream API manufacturers.
The Novel Approach
The innovative approach detailed in the patent data circumvents these issues by fundamentally altering the solvent environment surrounding the product molecules. Instead of attempting to crystallize from the original reaction medium, the process involves transferring the crude mixture into a high-boiling polar solvent system. This strategic shift exploits the specific interactions between the solvent molecules and the impurities. The high-boiling polar solvent acts as a selective trap for unwanted byproducts through mechanisms such as hydrogen bonding, effectively keeping them in solution while the desired product precipitates. This method allows for the removal of low-boiling non-polar solvents under mild vacuum conditions, preventing thermal degradation of the sensitive chiral intermediate. The result is a streamlined workflow that converts difficult-to-handle oils into high-purity crystals with minimal operational complexity, offering a clear path toward cost-effective and scalable manufacturing.
Mechanistic Insights into Solvent Exchange Crystallization
The core mechanism driving the success of this purification lies in the differential solvation effects exerted by protic polar solvents on the various components of the reaction mixture. When the mother liquor containing the reduced product is introduced to solvents like methanol, ethanol, or isopropanol, a dynamic equilibrium is established. The impurities, which often contain functional groups capable of acting as hydrogen bond acceptors or donors, interact strongly with the hydroxyl groups of the polar solvent. This interaction creates a solvation shell around the impurity molecules, significantly increasing their solubility in the polar phase compared to the target product. Conversely, the Ezetimibe intermediate, with its specific steric and electronic configuration, reaches its saturation point more rapidly under these conditions. This divergence in solubility behavior is the key to the high separation efficiency observed, allowing for the isolation of the product with an ee value surpassing 99 percent.
Furthermore, the process meticulously controls the thermodynamic parameters to optimize crystal growth and impurity exclusion. The removal of the low-boiling solvent is conducted at temperatures ranging from 5 degrees Celsius to 30 degrees Celsius under reduced pressure. This gentle evaporation prevents the co-precipitation of impurities that might occur during rapid solvent removal or high-temperature distillation. Following the concentration step, the system is cooled to sub-zero temperatures, typically between 0 degrees Celsius and minus 20 degrees Celsius. This controlled cooling regime promotes the formation of a stable crystal lattice that inherently rejects impurity molecules, a phenomenon known as lattice exclusion. The combination of specific solvent selection and precise thermal management ensures that the final product not only meets chemical purity specifications but also possesses the correct polymorphic form and particle size distribution required for subsequent processing steps.
How to Synthesize Ezetimibe Intermediate Efficiently
The synthesis and purification workflow described herein offers a practical blueprint for manufacturing teams aiming to implement this technology. The process begins with the crude mother liquor obtained from the chiral reduction of the ketone precursor using agents such as dimethyl sulfide borane complex or (-)-DIP-Cl. Rather than working up the reaction in the traditional manner, the entire mixture is directly subjected to the solvent exchange protocol. This eliminates the need for intermediate isolation steps that often lead to material loss. The detailed standardized synthesis steps below outline the precise ratios, temperatures, and operational sequences required to replicate the high yields and purity reported in the patent data. Adhering to these parameters is crucial for maintaining the integrity of the chiral center and ensuring consistent batch-to-batch quality.
- Transfer the mother liquor containing the reduced product in a low-boiling solvent into a high-boiling polar solvent system.
- Concentrate the mixture under reduced pressure at mild temperatures to remove the volatile non-polar solvent components.
- Induce crystallization by cooling the remaining polar solution to sub-zero temperatures, followed by filtration and vacuum drying.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this purification technology translates into tangible strategic benefits beyond mere technical specifications. The simplification of the purification process directly impacts the cost structure of the intermediate by reducing the number of unit operations and the consumption of auxiliary materials. By avoiding complex chromatographic columns and extensive solvent swaps, manufacturers can significantly lower their operational expenditures. The robustness of the method also means that variations in the quality of the upstream crude material can be tolerated without compromising the final specification, thereby reducing the risk of batch failures. This reliability is paramount for maintaining continuous supply lines to API production facilities, where interruptions can have cascading effects on drug availability. The ability to consistently deliver high-purity material with minimal environmental footprint further aligns with the sustainability goals of modern pharmaceutical enterprises.
- Cost Reduction in Manufacturing: The elimination of expensive chromatographic purification steps and the reduction in solvent usage volumes lead to substantial cost savings in the overall manufacturing process. By utilizing common, industrially available solvents like methanol and ethyl acetate, the process avoids the need for specialized or hazardous reagents that drive up procurement costs. The high yield achieved, often exceeding 90 percent, ensures that raw material utilization is maximized, further driving down the cost per kilogram of the final intermediate. Additionally, the mild reaction conditions reduce energy consumption associated with heating and cooling, contributing to a leaner and more economical production model that enhances competitiveness in the global market.
- Enhanced Supply Chain Reliability: The simplicity and robustness of this purification method significantly de-risk the supply chain for Ezetimibe intermediates. Because the process is less sensitive to minor fluctuations in reaction conditions compared to traditional methods, it ensures a higher success rate for production batches. This consistency allows suppliers to provide more accurate delivery forecasts and maintain safety stock levels with greater confidence. The use of standard equipment and widely available solvents means that production is not bottlenecked by the scarcity of specialized materials or machinery. Consequently, partners can rely on a steady flow of high-quality intermediates, minimizing the risk of production stoppages at the API manufacturing stage and ensuring uninterrupted availability of the final medication for patients.
- Scalability and Environmental Compliance: This purification route is inherently designed for scale-up, utilizing unit operations that are standard in fine chemical manufacturing facilities worldwide. The transition from laboratory scale to multi-ton commercial production is seamless, as the physics of solvent exchange and crystallization remain consistent across different vessel sizes. From an environmental perspective, the process generates less waste solvent and avoids the disposal issues associated with silica gel from chromatography. The ability to recover and recycle the high-boiling polar solvents further reduces the environmental impact, aligning with green chemistry principles. This scalability and compliance profile make the technology an attractive option for companies looking to expand their production capacity while adhering to increasingly strict regulatory and environmental standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this purification technology. They are derived from a detailed analysis of the patent specifications and are intended to clarify the operational advantages and mechanistic underpinnings of the process. Understanding these aspects is essential for technical teams evaluating the feasibility of adopting this method for their specific production lines. The answers provided reflect the empirical data and theoretical framework established in the intellectual property documentation, offering a clear perspective on the value proposition of this innovation.
Q: How does this purification method remove impurities effectively?
A: The method utilizes the differential solubility and hydrogen bonding capabilities of protic polar solvents. Impurities interact strongly with the polar solvent matrix, remaining in solution while the target product crystallizes out with high optical purity.
Q: What represents the key operational advantage of this process?
A: The process operates under mild conditions with simple unit operations like concentration and filtration, avoiding complex chromatographic separations and significantly reducing processing time and energy consumption.
Q: Is this method suitable for large-scale industrial production?
A: Yes, the technique relies on standard chemical engineering equipment such as reactors and centrifuges, making it highly scalable from pilot batches to multi-ton commercial manufacturing without losing yield or purity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ezetimibe Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-purity intermediates in the development and manufacture of life-saving cardiovascular medications. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless. We are equipped with rigorous QC labs and adhere to stringent purity specifications to guarantee that every batch of Ezetimibe intermediate meets the exacting standards required by global regulatory bodies. Our commitment to quality is matched by our dedication to process optimization, allowing us to deliver materials that facilitate efficient downstream processing for our clients.
We invite potential partners to engage with our technical procurement team to discuss how this advanced purification technology can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this refined manufacturing route. We encourage you to contact us to obtain specific COA data and route feasibility assessments tailored to your project requirements. Together, we can drive efficiency and quality in the production of essential pharmaceutical intermediates, ensuring a healthier future through superior chemical manufacturing.