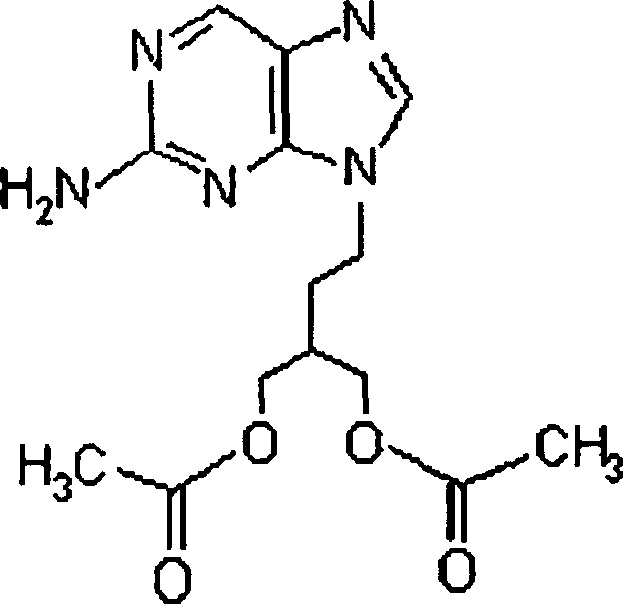
Revolutionizing Antiviral API Production with 100% Selective Purine Intermediates
The pharmaceutical industry continuously seeks robust synthetic pathways for antiviral agents, particularly for nucleoside analogs like famciclovir, which serve as critical prodrugs in treating herpes virus infections. Patent CN1791602A introduces a groundbreaking methodology centered on a novel class of compounds, specifically 2-amino-9-(2-substituted ethyl)purines, represented by general formula (II'). This intellectual property represents a significant leap forward in process chemistry by addressing long-standing issues regarding regioselectivity and safety in purine alkylation. By leveraging these new intermediates, manufacturers can achieve a theoretical 100% selectivity for the desired N-9 alkylated product, effectively bypassing the formation of pharmacologically inactive N-7 isomers that plague traditional synthesis routes. The strategic implementation of this technology allows for the production of high-purity 9-[4-acetoxy-3-(acetoxymethyl)butan-1-yl]-2-aminopurine under remarkably mild reaction conditions, thereby enhancing overall process efficiency and operational safety profiles for global supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
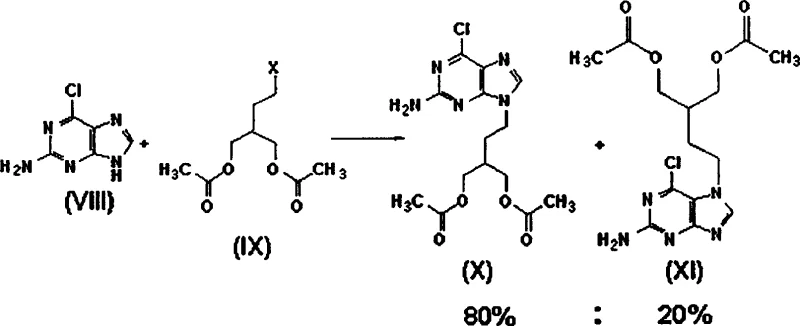
The Limitations of Conventional Methods
Historically, the industrial synthesis of famciclovir has been hindered by inherent chemical inefficiencies and significant safety hazards associated with catalytic hydrogenation. Prior art, such as European Patent No. 182,024 and U.S. Patent No. 5,684,153, describes reacting 2-amino-6-chloropurine with halogenated side chains, a process that notoriously yields a mixture of regioisomers. As illustrated in the reaction schemes below, the alkylation step typically produces the desired 9-substituted compound alongside its 7-substituted isomer in a ratio of approximately 80% to 20%, necessitating complex and costly purification steps to isolate the active pharmaceutical ingredient. Furthermore, subsequent conversion to the final drug often relies on palladium-catalyzed reduction under high pressure exceeding 50 psi, introducing severe explosion risks and requiring specialized, expensive high-pressure reactor infrastructure that limits scalability and increases capital expenditure for manufacturing facilities.
The Novel Approach
In stark contrast to these legacy methods, the innovative pathway disclosed in CN1791602A fundamentally restructures the synthetic logic by utilizing a pre-functionalized purine core that dictates regioselectivity from the outset. Instead of attempting to control alkylation on a bare purine ring where N-7 and N-9 competition is inevitable, this method employs 2-amino-9-(2-haloethyl)purine derivatives as the key starting materials. This strategic shift ensures that the side chain is already attached at the correct N-9 position before the construction of the acyclic sugar moiety begins, thereby guaranteeing 100% selectivity and eliminating the formation of the inactive N-7 isomer entirely. Additionally, the replacement of hazardous high-pressure hydrogenation with standard chemical reduction techniques using reagents like sodium borohydride allows the entire process to proceed at atmospheric pressure and moderate temperatures, significantly lowering the barrier to entry for safe, large-scale manufacturing while simultaneously reducing the environmental footprint associated with heavy metal catalyst disposal.
Mechanistic Insights into Regioselective N-Alkylation and Cyclization
The core mechanistic advantage of this technology lies in the stability and reactivity profile of the 2-amino-9-(2-substituted ethyl)purine intermediate, specifically when the substituent is a halogen such as bromine. In traditional approaches, the nucleophilic attack on the purine ring is governed by thermodynamic and kinetic factors that often favor a mixture of products; however, by pre-installing the ethyl linker at the N-9 position, the electronic environment of the ring is modified to prevent subsequent migration or competing alkylation at the N-7 nitrogen. The subsequent reaction with diethyl malonate proceeds via a clean nucleophilic substitution mechanism where the terminal halogen of the ethyl side chain is displaced by the carbanion of the malonate, extending the carbon chain with high fidelity. This step is critical as it builds the requisite four-carbon backbone needed for the final acyclic nucleoside structure without introducing chiral centers that would require resolution, thus streamlining the synthetic pathway and maximizing atom economy throughout the transformation sequence.
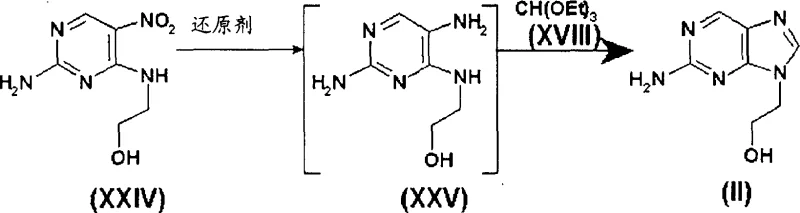
Impurity control is another pivotal aspect where this novel mechanism outperforms conventional chemistry, particularly regarding the exclusion of Formula XXIII, the 7-isomer of famciclovir which possesses no medicinal value. In the new process, the starting material 2-amino-9-(2-hydroxyethyl)purine is inherently free of its 7-isomer counterpart, 2-amino-7-(2-hydroxyethyl)purine, due to the specific cyclization conditions used to generate it from 2-amino-4-(2-hydroxyethylamino)-5-nitropyrimidine. This purity is carried through the halogenation and chain-extension steps, meaning that the final acetylation reaction yields exclusively the target molecule without the burden of isomeric contamination. From a quality control perspective, this simplifies the analytical workload and ensures that the final API meets stringent regulatory specifications for related substances without the need for repetitive recrystallization or chromatographic separation, which are often the primary drivers of yield loss in nucleoside manufacturing.
How to Synthesize 9-[4-Acetoxy-3-(acetoxymethyl)butan-1-yl]-2-aminopurine Efficiently
The practical execution of this synthesis involves a streamlined four-step sequence that transforms simple nitropyrimidine precursors into the complex famciclovir structure with exceptional efficiency. The process begins with the preparation of the key 2-amino-9-(2-hydroxyethyl)purine intermediate, followed by halogenation to activate the side chain for nucleophilic attack. Detailed operational parameters, including specific solvent systems like dimethyl sulfoxide or acetonitrile and precise temperature controls ranging from 20°C to 60°C, are critical to maintaining the high yields reported in the patent examples. The following guide outlines the standardized protocol derived from the experimental data, ensuring reproducibility and safety for process chemists aiming to implement this superior route in a GMP environment.
- Halogenate 2-amino-9-(2-hydroxyethyl)purine using agents like dibromotriphenylphosphine to form 2-amino-9-(2-haloethyl)purine with high yield.
- React the halogenated intermediate with diethyl malonate in the presence of a base such as potassium carbonate to extend the carbon chain.
- Reduce the diester intermediate using sodium borohydride or lithium aluminum hydride to generate the diol precursor.
- Acetylate the diol using acetic anhydride under mild conditions to finalize the synthesis of 9-[4-acetoxy-3-(acetoxymethyl)butan-1-yl]-2-aminopurine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers transformative benefits that extend far beyond simple chemical yield improvements. By fundamentally altering the reaction mechanism to avoid hazardous high-pressure steps and toxic heavy metal catalysts, the process significantly de-risks the manufacturing operation, leading to lower insurance premiums and reduced regulatory compliance burdens associated with handling explosive materials. The elimination of palladium not only removes a volatile cost component subject to market fluctuations but also eradicates the need for expensive metal scavenging resins and the complex waste treatment protocols required to meet residual metal limits in pharmaceutical products, thereby driving down the total cost of goods sold substantially.
- Cost Reduction in Manufacturing: The removal of the palladium catalyst and high-pressure hydrogenation equipment results in drastic capital expenditure savings and lower operational costs. Without the need for specialized high-pressure reactors or expensive noble metal catalysts, the process becomes accessible to a wider range of contract manufacturing organizations, fostering competitive pricing. Furthermore, the 100% selectivity eliminates the yield losses associated with separating isomeric impurities, ensuring that nearly all raw material input is converted into saleable product, which directly enhances profit margins and reduces the cost per kilogram of the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as diethyl malonate, sodium borohydride, and acetic anhydride ensures a robust and stable supply chain that is not vulnerable to the geopolitical or logistical disruptions often seen with specialized catalysts. The mild reaction conditions allow for production in standard stainless steel reactors available in most multipurpose chemical plants, increasing the pool of qualified suppliers and reducing lead times for batch production. This flexibility enables manufacturers to respond more agilely to market demand surges for antiviral medications without being bottlenecked by limited access to high-pressure processing capabilities or scarce catalytic materials.
- Scalability and Environmental Compliance: Scaling this process from pilot plant to commercial tonnage is straightforward due to the absence of exothermic high-pressure hydrogenation steps that pose safety challenges at large volumes. The use of common organic solvents and the generation of non-toxic byproducts simplify effluent treatment, aligning with increasingly stringent global environmental regulations regarding heavy metal discharge. This green chemistry profile not only facilitates faster regulatory approval in key markets but also supports corporate sustainability goals by minimizing the environmental footprint of the manufacturing process, making it an attractive option for eco-conscious pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel famciclovir synthesis route. These answers are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this technology resolves specific pain points found in legacy manufacturing methods. Understanding these distinctions is crucial for technical teams evaluating process transfers and for strategic planners assessing the long-term viability of this supply source.
Q: How does this new method improve selectivity compared to conventional palladium-catalyzed routes?
A: Conventional methods often produce a mixture of N-9 and N-7 isomers with ratios like 80:20 or 94:6, requiring difficult purification. This novel route utilizes a pre-formed 2-amino-9-(2-substituted ethyl)purine intermediate which ensures 100% selectivity for the N-9 position, completely eliminating the formation of the inactive N-7 isomer (Formula XXIII).
Q: What are the safety advantages of avoiding palladium catalysts in famciclovir production?
A: Traditional synthesis requires highly explosive palladium catalysts under high pressure (over 50 psi), posing significant industrial safety risks. The new method replaces this hazardous reduction step with standard chemical reductions using agents like sodium borohydride under mild atmospheric conditions, drastically improving process safety and lowering insurance and compliance costs.
Q: Can this synthesis route be scaled for commercial API manufacturing?
A: Yes, the process is designed for high industrial efficiency. It operates at mild temperatures (20-60°C for key steps) and avoids high-pressure equipment. The elimination of heavy metal catalysts also simplifies waste treatment and purification, making it highly suitable for large-scale commercial production of antiviral intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Famciclovir Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to superior synthetic routes requires a partner with deep technical expertise and proven manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent can be fully realized in a practical, industrial setting. We maintain stringent purity specifications and operate rigorous QC labs equipped to detect trace impurities, guaranteeing that every batch of 2-amino-9-(2-substituted ethyl)purine or final famciclovir intermediate meets the highest global pharmacopeial standards for antiviral drug manufacturing.
We invite you to engage with our technical procurement team to discuss how this innovative chemistry can be integrated into your supply chain to drive efficiency and reduce costs. By requesting a Customized Cost-Saving Analysis, you can gain specific insights into the economic impact of switching to this palladium-free route for your specific volume requirements. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions based on hard data and our commitment to delivering high-quality pharmaceutical intermediates reliably.