Advanced Synthesis of Avibactam Sodium Intermediate for Commercial Scale-up
Introduction to Patent CN111646991A
The pharmaceutical industry continuously seeks robust and scalable pathways for critical beta-lactamase inhibitors, and patent CN111646991A presents a significant breakthrough in the preparation of Avibactam sodium intermediates. This intellectual property discloses a highly efficient method for synthesizing the key intermediate Compound (VII), which serves as the foundational scaffold for the final active pharmaceutical ingredient. Unlike traditional approaches that suffer from complex protection-deprotection sequences, this innovation leverages a strategic tert-butyl substitution to streamline the cyclization process. The structural integrity of the final Avibactam sodium molecule relies heavily on the stereochemical purity of this intermediate, making the synthetic route chosen here paramount for downstream efficacy.
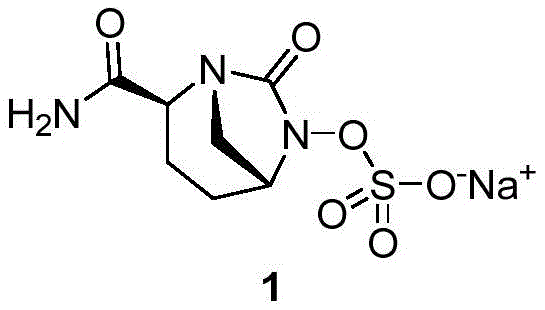
Furthermore, the disclosed methodology addresses the critical need for industrial viability by utilizing readily available starting materials and avoiding hazardous high-pressure operations. For R&D directors and procurement specialists, understanding the nuances of this patent is essential for securing a reliable supply chain of high-purity API intermediates. The transition from laboratory-scale curiosity to commercial reality hinges on the ability to control impurities and maximize atom economy, both of which are central themes in this technical disclosure. By focusing on the conversion of Compound (V) to Compound (VII) through a novel cyclization and deprotection sequence, the patent offers a compelling alternative to legacy manufacturing processes.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Avibactam sodium precursors has been plagued by inefficient multi-step sequences that negatively impact both cost and throughput. As illustrated in prior art documents such as CN102056901B, conventional Route 1 requires the protection of the piperidine nitrogen with expensive reagents like Fmoc-Cl before cyclization can occur. This additional step is not only costly but also introduces significant yield losses, often dropping below 50% during the ring-closing phase due to competing side reactions between the amide and the unprotected amine. Moreover, these legacy routes frequently rely on hydrogenation for debenzylation, a process that poses severe safety risks and scalability challenges when moving from kilogram to ton-scale production.
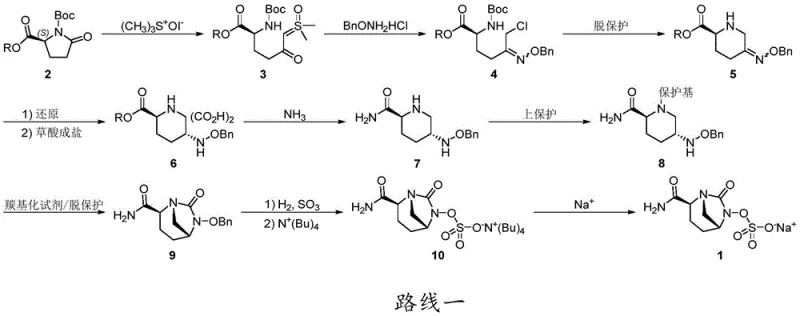
In addition to the economic burdens, the reliance on column chromatography for purification in older methods creates a bottleneck for continuous manufacturing. The use of unstable ester intermediates in Route 2 further complicates the landscape, often requiring autoclave conditions or expensive enzymatic catalysis to achieve acceptable conversion rates. These technical hurdles result in a fragmented supply chain where the availability of high-purity intermediates is inconsistent. For supply chain heads, the inability to predictably scale these complex routes translates into extended lead times and increased inventory costs, making the search for a more direct synthetic pathway a strategic imperative for any organization aiming to dominate the beta-lactamase inhibitor market.
The Novel Approach
The innovative strategy outlined in patent CN111646991A fundamentally reimagines the synthetic logic by introducing a tert-butyl substituent on the nitrogen atom of the precursor Compound (V). This structural modification allows for direct cyclization using carbonylation reagents without the need for prior amine protection, effectively bypassing the yield-killing side reactions observed in conventional methods. The resulting bridged ring Compound (VI) is formed with exceptional efficiency, boasting yields exceeding 85%, which represents a dramatic improvement over the sub-50% yields of the past. This streamlined approach not only simplifies the operational workflow but also enhances the overall atom economy of the process, aligning perfectly with modern green chemistry principles.
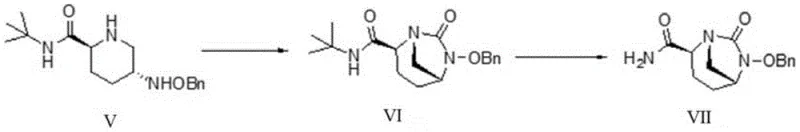
Moreover, the deprotection step in this new route utilizes mild Lewis acid conditions rather than dangerous hydrogenolysis, significantly reducing the operational hazards associated with high-pressure hydrogen gas. The use of boron trifluoride complexes facilitates the clean removal of the tert-butyl group under controlled temperatures, ensuring that the sensitive bicyclic core remains intact. This methodological shift enables the production of Compound (VII) with superior chiral selectivity and minimal diastereomeric impurities. For manufacturers, this means a more robust process that is easier to validate and scale, providing a distinct competitive advantage in the production of cost-effective and high-quality pharmaceutical intermediates.
Mechanistic Insights into Carbonylation and Acid-Mediated Deprotection
The core of this technological advancement lies in the precise mechanistic execution of the cyclization reaction, where Compound (V) undergoes intramolecular ring closure to form the diazabicyclooctanone skeleton. The reaction is initiated by the activation of the secondary amine through nucleophilic attack on a carbonylation reagent, such as triphosgene or N,N'-carbonyldiimidazole (CDI), in the presence of a mild base like sodium bicarbonate or triethylamine. This activation generates a reactive carbamoyl intermediate that spontaneously cyclizes onto the adjacent hydroxylamine moiety, forging the critical urea linkage within the bridged ring system. The choice of solvent, typically dichloromethane or ethyl acetate, plays a crucial role in stabilizing the transition state and preventing the formation of oligomeric byproducts, thereby ensuring high conversion rates.
Following the successful construction of the bicyclic framework, the deprotection mechanism employs a Lewis acid-catalyzed cleavage of the tert-butyl carbamate bond. Unlike protonic acids which might induce unwanted hydrolysis of the lactam ring, Lewis acids such as boron trifluoride acetic acid complex coordinate specifically with the carbonyl oxygen, facilitating the generation of a stable tert-butyl cation and the release of carbon dioxide. This gentle yet effective cleavage preserves the stereochemical integrity of the chiral centers at the 2S and 5R positions, which is vital for the biological activity of the final drug substance. The rigorous control of reaction temperature and stoichiometry during this phase ensures that impurity profiles remain well within specification limits, delivering a product suitable for direct progression to the final sulfation steps.
How to Synthesize Avibactam Sodium Intermediate Efficiently
The practical implementation of this synthesis requires strict adherence to the optimized reaction parameters detailed in the patent examples to ensure reproducibility and safety. The process begins with the preparation of the oxalate salt of Compound (V), which serves as a stable and purifiable intermediate that locks in the desired stereochemistry before the critical ring-closing step. Operators must maintain precise temperature controls during the reduction and salt formation phases to minimize the generation of diastereomers, as even minor deviations can compromise the optical purity of the final batch. Once the precursor is secured, the cyclization is performed under inert atmosphere conditions to prevent moisture interference with the sensitive carbonylation reagents.
- Prepare Compound V or its oxalate salt by reacting protected pyroglutamic acid derivatives with trimethyl sulfoxide iodide, followed by benzyloxyamine substitution and reduction.
- Perform cyclization of Compound V using a carbonylation reagent such as triphosgene or CDI in the presence of a base to form the bridged ring structure of Compound VI.
- Execute deprotection of Compound VI using a Lewis acid like boron trifluoride complex to remove the tert-butyl group, yielding the final key intermediate Compound VII.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this novel synthetic route offers transformative benefits for procurement managers seeking to optimize the cost structure of API manufacturing. By eliminating the requirement for expensive protecting group reagents like Fmoc-Cl and removing the need for resource-intensive column chromatography purification, the overall cost of goods sold is significantly reduced. The process relies on commodity chemicals and standard unit operations such as crystallization and filtration, which are far more economical and easier to source globally than specialized catalysts or high-pressure equipment. This simplification of the bill of materials directly translates to improved margin potential and greater price stability for the finished intermediate.
- Cost Reduction in Manufacturing: The streamlined nature of the synthesis removes multiple low-yield steps that traditionally erode profitability in complex organic synthesis. By achieving higher yields in the cyclization step and avoiding the capital expenditure associated with hydrogenation reactors, manufacturers can realize substantial cost savings per kilogram of output. Furthermore, the ability to telescope multiple steps without isolating unstable intermediates reduces solvent consumption and waste disposal costs, contributing to a leaner and more financially efficient production model that enhances competitiveness in the global marketplace.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials and the avoidance of specialized high-pressure infrastructure mitigate many common supply chain risks. Since the process does not depend on scarce enzymes or custom-synthesized protecting groups, the risk of raw material shortages is drastically minimized. This robustness ensures consistent delivery schedules and reduces the likelihood of production stoppages due to equipment maintenance or reagent availability, providing pharmaceutical partners with a dependable source of critical intermediates that supports uninterrupted drug manufacturing timelines.
- Scalability and Environmental Compliance: The replacement of hazardous hydrogenation steps with mild acid-mediated deprotection significantly lowers the safety profile of the plant, facilitating easier regulatory approval for scale-up. The use of greener solvents and the reduction of heavy metal or toxic reagent usage align with increasingly stringent environmental regulations, reducing the burden of waste treatment and compliance reporting. This environmental stewardship not only protects the corporate reputation but also future-proofs the manufacturing site against evolving regulatory landscapes, ensuring long-term operational continuity and sustainability.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on why this route represents a superior choice for industrial application. Understanding these details helps stakeholders make informed decisions about technology transfer and process validation.
Q: Why does this new method avoid hydrogenation for deprotection?
A: Traditional routes often use N-benzyl protecting groups which require dangerous high-pressure hydrogenation for removal. This patent utilizes a tert-butyl group that can be removed under mild acidic conditions, significantly improving safety and scalability.
Q: How does this process improve yield compared to prior art?
A: By eliminating the need for additional amine protection steps (like Fmoc-Cl) before cyclization, the new route reduces side reactions and operational steps, achieving yields over 85% for the cyclization step compared to less than 50% in older methods.
Q: What represents the key cost-saving advantage of this synthesis?
A: The process avoids expensive reagents like Fmoc-Cl and eliminates the need for column chromatography purification, relying instead on crystallization. This drastically reduces raw material costs and processing time.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Avibactam Sodium Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic methodologies to meet the rigorous demands of the global pharmaceutical market. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patent CN111646991A are fully realized in practical manufacturing environments. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of Avibactam sodium intermediate meets the highest standards of quality and consistency required by regulatory bodies worldwide.
We invite forward-thinking organizations to collaborate with us to leverage this cutting-edge technology for their supply chains. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and gain access to specific COA data and route feasibility assessments. Let us partner with you to secure a sustainable, cost-effective, and high-quality supply of this vital pharmaceutical intermediate, driving your projects forward with confidence and reliability.