Advanced Catalytic Ring-Opening of Chromones for High-Purity Cyano Pharmaceutical Intermediates
The strategic incorporation of cyano functional groups into small molecule scaffolds remains a cornerstone of modern medicinal chemistry, driven by the group's unique ability to enhance binding affinity through hydrogen bonding and improve metabolic stability. Patent CN111848451B introduces a transformative methodology for accessing these critical motifs via the ring-opening cyanation of chromone derivatives. This innovation addresses long-standing challenges in the synthesis of (E)-4-(2-hydroxyphenyl)-4-oxobut-2-enenitrile structures, which serve as versatile building blocks for complex heterocyclic frameworks. By leveraging a metal-free Lewis acid catalytic system, the disclosed process offers a streamlined pathway that aligns perfectly with the rigorous purity standards demanded by global pharmaceutical supply chains. As a leader in fine chemical manufacturing, we recognize this technology as a pivotal advancement for producing high-purity pharmaceutical intermediates with exceptional efficiency and scalability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes to cyano-substituted chalcones and related enones often rely heavily on transition metal catalysis or harsh stoichiometric reagents that complicate the manufacturing landscape. Conventional methods frequently necessitate the use of expensive palladium or copper catalysts, which not only inflate raw material costs but also introduce significant regulatory hurdles regarding heavy metal residues in final drug substances. Furthermore, many established protocols require extreme reaction conditions, such as high temperatures or strong bases, which can lead to poor chemoselectivity and the formation of difficult-to-remove polymeric byproducts. These inefficiencies result in prolonged processing times, complex workup procedures involving extensive scavenging steps, and ultimately, reduced overall yields that strain production capacity. For procurement and supply chain managers, these factors translate into unpredictable lead times and elevated cost structures that hinder the commercial viability of potential drug candidates.
The Novel Approach
In stark contrast, the methodology detailed in CN111848451B utilizes tris(pentafluorophenyl)borane, a potent and selective Lewis acid, to facilitate the ring-opening of chromones with trimethylsilyl cyanide (TMSCN). This approach operates under remarkably mild conditions, typically requiring temperatures between 60°C and 80°C in common solvents like toluene. The reaction proceeds with high atom economy and exceptional functional group tolerance, accommodating a wide array of substituents including halogens, nitro groups, and alkyl chains without the need for protecting groups. 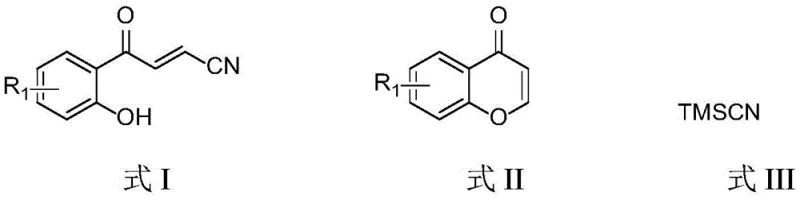 By eliminating transition metals entirely, this novel route drastically simplifies the purification process, often requiring only standard column chromatography to achieve analytical purity. The robustness of this catalytic system ensures consistent performance across diverse substrates, making it an ideal candidate for the reliable supply of complex pharmaceutical intermediates on a commercial scale.
By eliminating transition metals entirely, this novel route drastically simplifies the purification process, often requiring only standard column chromatography to achieve analytical purity. The robustness of this catalytic system ensures consistent performance across diverse substrates, making it an ideal candidate for the reliable supply of complex pharmaceutical intermediates on a commercial scale.
Mechanistic Insights into B(C6F5)3-Catalyzed Ring-Opening Cyanation
The efficacy of this transformation hinges on the unique Lewis acidity of tris(pentafluorophenyl)borane, which activates the carbonyl oxygen of the chromone substrate towards nucleophilic attack. Upon coordination, the electron density at the carbonyl carbon is significantly depleted, rendering the adjacent carbon-oxygen bond susceptible to cleavage by the cyanide source. Trimethylsilyl cyanide serves as a safe and manageable cyanide equivalent, where the silicon center interacts with the boron-activated species to deliver the cyanide ion selectively. This mechanism avoids the generation of free cyanide salts, enhancing operational safety while maintaining high reactivity. The subsequent rearrangement leads to the formation of the stable enone nitrile structure with exclusive E-selectivity, a critical stereochemical feature for downstream biological activity. Understanding this mechanistic pathway allows process chemists to fine-tune reaction parameters for optimal throughput and minimal impurity formation.
Impurity control is inherently superior in this metal-free system due to the absence of redox-active species that often trigger side reactions such as homocoupling or over-reduction. The mild thermal profile prevents thermal degradation of sensitive functional groups, ensuring that the integrity of the molecular scaffold is preserved throughout the synthesis. For instance, substrates bearing electron-withdrawing groups like nitro or trifluoromethyl moieties, which might be unstable under strongly basic conditions, remain intact and react smoothly. 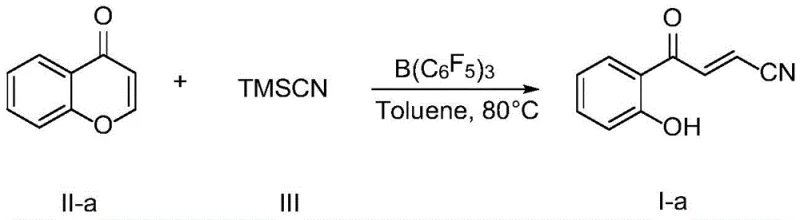 This high level of chemoselectivity minimizes the formation of closely related structural impurities, thereby reducing the burden on analytical quality control laboratories. The result is a cleaner crude product profile that facilitates easier isolation and higher final purity, directly addressing the stringent specifications required for active pharmaceutical ingredient (API) precursors.
This high level of chemoselectivity minimizes the formation of closely related structural impurities, thereby reducing the burden on analytical quality control laboratories. The result is a cleaner crude product profile that facilitates easier isolation and higher final purity, directly addressing the stringent specifications required for active pharmaceutical ingredient (API) precursors.
How to Synthesize (E)-4-(2-hydroxyphenyl)-4-oxobut-2-enenitrile Efficiently
The practical implementation of this synthesis is straightforward and amenable to standard laboratory and pilot plant equipment. The process begins by charging a reaction vessel, such as a Schlenk flask, with the chosen chromone derivative and the tris(pentafluorophenyl)borane catalyst under an inert nitrogen atmosphere to exclude moisture. Dry toluene is added to establish a molar concentration of the chromone substrate between 0.1 and 0.3 mol/L, ensuring optimal solubility and reaction kinetics. Trimethylsilyl cyanide is then introduced in a slight molar excess, typically ranging from 1.2 to 2.4 equivalents, to drive the equilibrium towards completion. The detailed standardized synthesis steps for scaling this reaction are provided in the guide below.
- Charge a Schlenk flask with chromone substrate, trimethylsilyl cyanide (TMSCN), and B(C6F5)3 catalyst under nitrogen protection.
- Add dry toluene solvent to achieve a concentration of 0.1-0.3 mol/L and heat the mixture to 80°C.
- Stir the reaction for 6 to 24 hours, then dilute with acetone, concentrate, and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this catalytic ring-opening strategy offers profound advantages that resonate deeply with the priorities of procurement managers and supply chain directors. The elimination of precious metal catalysts removes a major cost driver and supply bottleneck, as the price volatility of metals like palladium is a well-known risk factor in chemical manufacturing. Additionally, the use of commodity chemicals such as toluene and TMSCN ensures that raw material sourcing is stable and geographically diverse, mitigating the risk of supply disruptions. The simplified workup procedure reduces solvent consumption and waste generation, aligning with increasingly strict environmental regulations and sustainability goals. These factors collectively contribute to a more resilient and cost-effective supply chain for critical pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The substitution of expensive transition metal catalysts with a reusable or low-loading organoboron catalyst results in substantial cost savings per kilogram of product. By avoiding the need for specialized metal scavengers and extensive purification trains required to meet residual metal limits, the overall processing cost is drastically simplified. This economic efficiency allows for more competitive pricing models without compromising on the quality or purity of the final intermediate, providing a clear financial advantage in high-volume production scenarios.
- Enhanced Supply Chain Reliability: The reliance on readily available and stable reagents ensures a consistent supply of key starting materials, reducing the lead time for high-purity pharmaceutical intermediates. Unlike processes dependent on specialized ligands or air-sensitive metal complexes, this method utilizes robust chemistry that is less prone to batch-to-batch variability caused by reagent degradation. This reliability translates into predictable production schedules and the ability to respond rapidly to fluctuating market demands from downstream API manufacturers.
- Scalability and Environmental Compliance: The reaction conditions are inherently scalable, utilizing standard heating and stirring equipment found in most multipurpose chemical plants. The absence of toxic heavy metals simplifies waste treatment protocols, lowering the environmental footprint and associated disposal costs. This ease of scale-up from gram to ton quantities ensures that the technology can support the entire lifecycle of a drug candidate, from early-stage clinical trials to full commercial manufacturing, without the need for disruptive process redesigns.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this chromone ring-opening technology. These insights are derived directly from the experimental data and scope defined in the patent literature, providing a factual basis for evaluating its suitability for your specific project needs. We encourage technical teams to review these details to understand the full potential of this synthetic route.
Q: What is the primary advantage of using B(C6F5)3 over transition metal catalysts?
A: The use of tris(pentafluorophenyl)borane eliminates the need for expensive and toxic transition metals like palladium or copper, significantly simplifying downstream purification and reducing heavy metal residue risks in pharmaceutical intermediates.
Q: What is the substrate scope for this ring-opening cyanation reaction?
A: The method demonstrates excellent tolerance for various substituents on the chromone ring, including electron-donating groups like methyl and methoxy, as well as electron-withdrawing groups such as fluoro, chloro, nitro, and trifluoromethyl.
Q: What are the typical reaction conditions required for high yield?
A: Optimal results are achieved using dry toluene as the solvent at temperatures between 60°C and 80°C, with a catalyst loading of 5 to 10% relative to the chromone substrate.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cyano Compound Supplier
At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative technologies like this are seamlessly translated into industrial reality. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications for even the most complex organic molecules. We understand that the transition from bench-scale discovery to commercial supply requires a partner who can navigate both chemical complexity and regulatory compliance with equal expertise. Our commitment to quality assurance guarantees that every batch of cyano compound delivered meets the exacting standards required for global pharmaceutical applications.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis method can optimize your specific supply chain. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits tailored to your volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our capabilities can support your development timelines and commercial goals effectively.