Advanced Manufacturing of Tolterodine Intermediates via Efficient Lactone Opening
Advanced Manufacturing of Tolterodine Intermediates via Efficient Lactone Opening
The pharmaceutical landscape for treating overactive bladder disorders relies heavily on the consistent supply of high-quality antimuscarinic agents, with Tolterodine standing as a cornerstone therapy. As detailed in patent CN101445462B, a transformative synthetic methodology has been established that fundamentally alters the production economics and technical feasibility of this critical active pharmaceutical ingredient (API). This innovative approach departs from the convoluted multi-step sequences of the past, offering a streamlined pathway that begins with the strategic activation of a chromen-2-one lactone scaffold. By leveraging advanced organometallic activation and selective reduction techniques, this process delivers Tolterodine free base and its tartrate salt with superior efficiency. For global procurement teams and R&D directors, understanding this technological leap is essential for securing a reliable tolterodine intermediate supplier capable of meeting stringent regulatory and volume demands.
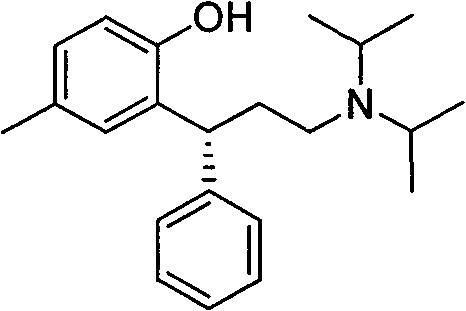
The structural complexity of Tolterodine, specifically the chiral benzylic position and the bulky diisopropylamine group, has historically posed significant challenges in stereoselective synthesis and impurity management. Traditional routes often struggled with racemization issues or required cumbersome resolution steps late in the synthesis, driving up costs and extending lead times. The methodology disclosed in CN101445462B addresses these pain points head-on by introducing a robust amide intermediate strategy. This not only simplifies the molecular construction but also enhances the overall stability of the process, making it an ideal candidate for the commercial scale-up of complex pharmaceutical intermediates. The ability to produce this molecule with fewer unit operations directly translates to reduced operational expenditure and a smaller environmental footprint, aligning perfectly with modern green chemistry initiatives in the fine chemical sector.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical precedents for Tolterodine synthesis, such as those outlined in U.S. Patent 5,382,600, reveal a laborious six-step trajectory that begins with trans-cinnamic acid and p-cresol. This archaic route necessitates a series of high-risk transformations, including methylation with methyl iodide, reduction of ester groups with lithium aluminum hydride, and a hazardous tosylation step followed by nucleophilic substitution. Each additional step in a synthetic sequence introduces a multiplicative loss in overall yield and a geometric increase in the potential for impurity generation. Furthermore, the reliance on boron tribromide for demethylation protection in later stages introduces severe safety hazards and corrosion issues that complicate reactor maintenance and waste disposal. Other methods, like those in patent WO2007147547, utilize cinnamyl chloride as a starting material; however, this precursor is notoriously expensive and difficult to source in bulk quantities, creating a bottleneck for cost reduction in API manufacturing. The cumulative effect of these inefficiencies is a process that is fragile, costly, and ill-suited for the high-volume demands of the global generic pharmaceutical market.
The Novel Approach
In stark contrast, the novel approach presented in the subject patent utilizes 3,4-dihydro-6-methyl-4-phenyl-2H-chromen-2-one (Compound 2) as a pivotal building block. This lactone precursor is not only more economically accessible but also possesses a reactive carbonyl functionality that can be directly manipulated to install the requisite amine side chain. The core innovation lies in the direct ring-opening amidation, where diisopropylamine reacts with the activated lactone to form Compound 3 in a single, high-yielding operation. This effectively collapses multiple synthetic steps into one, drastically shortening the critical path to the final API. By eliminating the need for etherification, demethylation, and complex protecting group strategies, the new route minimizes solvent consumption and waste generation. This streamlined architecture allows manufacturers to achieve high-purity pharmaceutical intermediates with significantly less downstream processing, ensuring that the final product meets the rigorous specifications required for human therapeutic use without the burden of excessive purification costs.
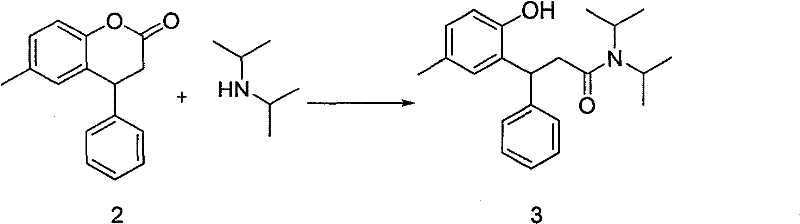
Mechanistic Insights into Lactone Activation and Amide Formation
The success of this synthetic route hinges on the precise activation of the lactone ring in Compound 2 to facilitate nucleophilic attack by diisopropylamine. The patent discloses several sophisticated activation strategies, ranging from the use of basic metal reagents like ethylmagnesium bromide (Grignard reagent) or butyllithium to the deployment of Lewis acids such as aluminum trichloride or titanium tetrachloride. In the Grignard-mediated pathway, the organomagnesium species likely coordinates with the lactone carbonyl oxygen, increasing the electrophilicity of the carbon center and promoting the formation of a tetrahedral intermediate. Upon quenching and acidification, this intermediate collapses to expel the phenolic oxygen (which remains part of the aromatic system) and establishes the stable amide bond found in Compound 3. Alternatively, the use of Lewis acids provides a milder yet highly effective activation mode, where the metal center coordinates with the carbonyl lone pairs, lowering the energy barrier for amine attack. This mechanistic flexibility is crucial for process chemists, as it allows for the optimization of reaction conditions based on available infrastructure and safety protocols, ensuring robust reducing lead time for high-purity pharmaceutical intermediates.
Following the formation of the amide intermediate, the subsequent reduction step (Step B) is equally critical for establishing the final amine functionality of Tolterodine. The patent outlines a versatile array of reducing agents, including lithium aluminum hydride (LiAlH4), sodium borohydride (NaBH4) combined with Lewis acids, and borane complexes. The reduction of tertiary amides to amines is a challenging transformation that typically requires potent hydride donors. LiAlH4 operates by delivering hydride ions to the amide carbonyl, forming an iminium ion intermediate that is subsequently reduced to the amine. However, the patent also highlights the utility of NaBH4 in the presence of Lewis acids like AlCl3 or Iodine, which activates the borohydride species to mimic the reactivity of more aggressive reagents while offering better handling characteristics. This diversity in reduction chemistry allows manufacturers to tailor the process to minimize the formation of over-reduced by-products or alcohol impurities, thereby maintaining a clean impurity profile that simplifies the final crystallization of the tartrate salt.
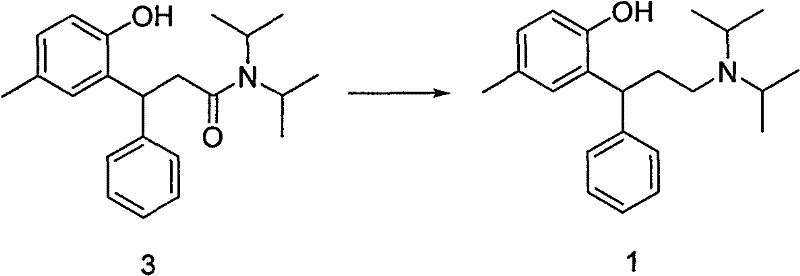
How to Synthesize Tolterodine Efficiently
The execution of this synthesis requires careful attention to stoichiometry, temperature control, and quenching procedures to maximize yield and safety. The process begins with the activation of the lactone under inert atmosphere, followed by the controlled addition of the amine source. Once the amide intermediate is isolated and purified, typically via crystallization from methanol, it undergoes reduction in a separate reactor. The final step involves the resolution or salt formation with L-(+) tartaric acid to secure the desired stereochemistry and physical properties of the API. Detailed standard operating procedures regarding specific molar ratios, solvent volumes, and thermal profiles are essential for reproducibility.
- Activate 3,4-dihydro-6-methyl-4-phenyl-2H-chromen-2-one (Compound 2) using a Grignard reagent, LDA, or Lewis acid, then react with diisopropylamine to form the amide intermediate (Compound 3).
- Reduce the amide intermediate (Compound 3) using a hydride reducing agent such as LiAlH4, Red-Al, or a NaBH4/Lewis acid combination to yield Tolterodine free base.
- Dissolve the Tolterodine free base in a suitable solvent and react with L-(+) tartaric acid to crystallize the final Tolterodine tartrate salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this lactone-based synthesis represents a strategic opportunity to de-risk the supply of Tolterodine. The primary economic driver here is the substantial simplification of the material bill. By removing the dependency on expensive and volatile starting materials like cinnamyl chloride or specialized brominated precursors, the overall cost of goods sold (COGS) is inherently lowered. The elimination of multiple isolation and purification steps between the starting material and the final amine further contributes to cost reduction in API manufacturing by reducing labor hours, solvent usage, and energy consumption. Additionally, the robustness of the amide intermediate allows for potential telescoping of steps, where the crude amide could potentially be carried forward without full isolation, although the patent describes a crystallization step for quality assurance. This flexibility enables suppliers to respond more agilely to market fluctuations and demand spikes.
- Cost Reduction in Manufacturing: The most significant financial benefit arises from the drastic shortening of the synthetic sequence. Conventional routes often involve six or more distinct chemical transformations, each incurring a yield loss and requiring separate workup procedures. By condensing the synthesis into essentially two major chemical transformations (amidation and reduction), the new method minimizes the accumulation of yield losses. Furthermore, the avoidance of hazardous reagents like boron tribromide reduces the capital expenditure required for specialized corrosion-resistant equipment and hazardous waste treatment facilities. The use of commodity chemicals such as diisopropylamine and common reducing agents ensures that raw material costs remain stable and predictable, shielding the supply chain from the price volatility associated with specialty fine chemicals.
- Enhanced Supply Chain Reliability: Supply continuity is paramount for life-saving medications, and this route enhances reliability by utilizing widely available starting materials. The precursor, 3,4-dihydro-6-methyl-4-phenyl-2H-chromen-2-one, can be sourced from multiple vendors or synthesized via well-established condensation reactions, preventing single-source bottlenecks. Unlike methods relying on chiral pool starting materials that may have limited global capacity, this racemic synthesis followed by resolution (or asymmetric variants if adapted) offers a scalable foundation. The simplified post-treatment processes, involving standard extractions and crystallizations, mean that production can be easily transferred between different manufacturing sites without extensive re-validation, ensuring that reducing lead time for high-purity pharmaceutical intermediates becomes a tangible reality rather than just a goal.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this process offers a cleaner profile. The reduction in step count directly correlates to a reduction in the E-factor (mass of waste per mass of product). By avoiding the generation of halogenated waste streams associated with tosylation and demethylation steps, the facility's waste treatment load is significantly lightened. The ability to use safer reducing alternatives, such as sodium borohydride combinations instead of purely pyrophoric reagents in certain contexts, enhances operational safety. This alignment with green chemistry principles not only lowers compliance costs but also future-proofs the manufacturing process against tightening environmental regulations, making it a sustainable choice for long-term commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and claims within the patent documentation, providing a clear picture of the technology's capabilities and limitations for potential partners.
Q: What are the primary advantages of the lactone opening route over cinnamyl chloride methods?
A: The lactone opening route utilizes readily available chromen-2-one precursors, avoiding the high cost and supply instability associated with cinnamyl chloride. Furthermore, it eliminates the need for complex protection and deprotection sequences, significantly simplifying the purification workflow.
Q: How does this method improve impurity control compared to prior art?
A: By bypassing the methylation and subsequent demethylation steps found in older patents (like US5382600), this method reduces the formation of ether by-products and halogenated impurities. The direct conversion from lactone to amide allows for cleaner reaction profiles and easier crystallization of the intermediate.
Q: Is this process scalable for industrial production?
A: Yes, the process is designed for industrial suitability. It employs standard unit operations such as reflux, extraction, and crystallization. The use of versatile reducing agents (including safer NaBH4 combinations) allows for flexible scale-up from pilot plants to multi-ton commercial manufacturing without requiring exotic equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tolterodine Supplier
At NINGBO INNO PHARMCHEM, we recognize that the theoretical advantages of a patent must be translated into practical, large-scale reality to create value for our partners. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the efficiencies promised by the lactone-opening route are fully realized in our manufacturing suites. We maintain stringent purity specifications through our rigorous QC labs, utilizing advanced analytical techniques to monitor the critical amide intermediate and the final free base for any trace impurities. Our commitment to quality ensures that every batch of Tolterodine intermediate we supply meets the exacting standards required for downstream API formulation, providing you with a foundation of trust and reliability.
We invite you to collaborate with us to leverage this advanced synthetic technology for your supply chain. By partnering with NINGBO INNO PHARMCHEM, you gain access not just to a product, but to a comprehensive technical partnership. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our optimized manufacturing process can drive down your costs while securing your supply of this vital urological medication.