Advanced Triphosgene-Mediated Synthesis of Mirabegron Intermediate for Commercial Scale-Up
Advanced Triphosgene-Mediated Synthesis of Mirabegron Intermediate for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust and cost-efficient pathways for producing critical API intermediates, particularly for high-value drugs like Mirabegron, a beta-3 adrenergic receptor agonist used for overactive bladder treatment. A significant technological breakthrough in this domain is detailed in patent CN108727212B, which discloses a novel synthesis method for the key chiral intermediate (R)-2-hydroxy-N-(4-nitrophenylethyl)-2-phenylacetamide. This innovation addresses longstanding challenges in amide bond formation by replacing traditional, expensive coupling agents with a more economical triphosgene-mediated activation strategy. The structural complexity of Mirabegron requires precise stereochemical control, and the disclosed route ensures the retention of chirality while drastically simplifying the purification workflow. By shifting away from carbodiimide chemistry, manufacturers can achieve superior impurity profiles and operational efficiency. This report analyzes the technical merits and commercial implications of this advanced synthetic route for global supply chain stakeholders.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of the mirabegron intermediate (II) has relied heavily on direct condensation reactions that present significant economic and environmental drawbacks for large-scale manufacturing. As illustrated in the prior art, Method A utilizes 1-ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDCI) and N-hydroxybenzotriazole (HOBt) to couple R-mandelic acid with 4-nitrophenylethylamine hydrochloride. While effective on a small laboratory scale, this approach generates substantial amounts of urea byproducts that are notoriously difficult to remove, necessitating rigorous and wasteful post-treatment protocols including multiple water washes, acid washes, alkali washes, and hydrochloric acid washes. Furthermore, the reliance on EDCI and HOBt introduces high raw material costs, and the subsequent toluene recrystallization steps increase solvent consumption and toxicity risks. Alternatively, Method B employs trimethyl borate (B(OMe)3) and DIPEA, but this route suffers from lower condensation yields and inferior product purity, often capping at around 98.65%, which is insufficient for stringent pharmaceutical specifications without further extensive purification.

The Novel Approach
In stark contrast to these legacy methods, the novel approach described in the patent utilizes a two-step sequence initiated by the activation of R-mandelic acid with triphosgene. This strategy fundamentally alters the reaction landscape by generating a highly reactive lactide or mixed anhydride intermediate (Intermediate I) in situ, which then undergoes efficient acylation with 4-nitrophenylethylamine. This pathway completely circumvents the need for expensive peptide coupling reagents like EDCI and HOBt, thereby eliminating the associated urea impurities that complicate downstream processing. The operational simplicity is a major advantage; the reaction proceeds under mild conditions, typically between 15°C and 30°C for the activation step, and the workup involves straightforward phase separation and concentration rather than exhaustive washing cycles. This transition not only reduces the chemical footprint but also enhances the overall throughput of the manufacturing process, making it an ideal candidate for reliable pharmaceutical intermediates supplier operations aiming for leaner production models.
Mechanistic Insights into Triphosgene-Mediated Activation and Acylation
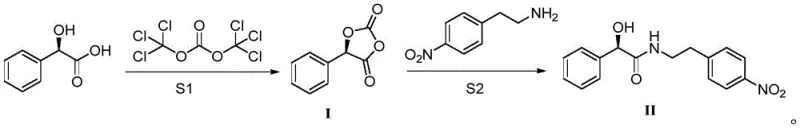
The core of this technological advancement lies in the mechanistic efficiency of using triphosgene as a dehydrating and activating agent for the carboxylic acid functionality of R-mandelic acid. In the first step (S1), triphosgene reacts with the mandelic acid in the presence of a base such as triethylamine or potassium carbonate within a polar aprotic solvent like acetonitrile or DMF. This reaction generates a reactive acyl chloride or mixed anhydride species, which rapidly cyclizes or stabilizes as Intermediate I. The use of triphosgene is particularly advantageous because it acts as a solid substitute for phosgene, offering safer handling characteristics while providing high atom economy for the activation step. The stoichiometry is carefully controlled, with a molar ratio of R-mandelic acid to triphosgene optimized at 1.0:1.1, ensuring complete conversion while minimizing excess reagent waste. This activation step is critical as it pre-organizes the electrophile for the subsequent nucleophilic attack, setting the stage for high-yield amide formation without racemization of the chiral center.
Following the formation of Intermediate I, the second step (S2) involves the nucleophilic attack by 4-nitrophenylethylamine to form the final amide bond. This acylation reaction is conducted in solvents such as acetone or acetonitrile at temperatures ranging from 40°C to 70°C, with 56°C identified as an optimal point for balancing reaction rate and selectivity. The mechanism ensures that the amine attacks the activated carbonyl carbon efficiently, displacing the leaving group generated from the triphosgene activation. Crucially, this pathway demonstrates exceptional impurity control, yielding a final product with a chemical purity of 99.73% and an isolated yield of 91%. The high purity is attributed to the clean nature of the triphosgene byproducts (HCl salts which are neutralized by the base) compared to the persistent urea derivatives formed in EDCI couplings. This mechanistic clarity provides R&D directors with confidence in the reproducibility and robustness of the process for high-purity pharmaceutical intermediates.
How to Synthesize (R)-2-hydroxy-N-(4-nitrophenylethyl)-2-phenylacetamide Efficiently
The implementation of this synthesis route requires precise control over reaction parameters to maximize the benefits of the triphosgene activation strategy. The process begins with the preparation of Intermediate I by reacting R-mandelic acid with triphosgene and a base in acetonitrile at 25°C for approximately 6 hours, monitored by TLC to ensure complete consumption of the starting acid. Once Intermediate I is isolated as a white solid with a near-quantitative yield of 99%, it is immediately subjected to acylation. The second stage involves dissolving Intermediate I in acetone and adding 4-nitrophenylethylamine, maintaining the temperature at 56°C for 3 hours. The final purification is remarkably simple, involving concentration followed by crystallization from methanol, which effectively removes any trace impurities. For a comprehensive understanding of the specific operational parameters and safety protocols, the detailed standardized synthesis steps are provided in the guide below.
- Perform esterification of R-mandelic acid with triphosgene in acetonitrile using triethylamine at 25°C to generate intermediate I.
- React intermediate I with 4-nitrophenylethylamine in acetone at 56°C to form the target amide bond.
- Purify the final product via methanol crystallization to achieve 99.73% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this triphosgene-based methodology represents a strategic opportunity to optimize cost structures and enhance supply reliability for mirabegron intermediate manufacturing. The elimination of high-cost coupling reagents like EDCI and HOBt directly translates to a significant reduction in raw material expenditure, as triphosgene is a commodity chemical with a much lower price point and higher availability. Furthermore, the simplified workup procedure, which avoids multiple aqueous washes and complex extractions, reduces the consumption of process water and organic solvents, leading to substantial cost savings in waste treatment and utility usage. The high yield of 91% and purity of 99.73% minimize the need for reprocessing or recycling off-spec material, thereby improving the overall material throughput and reducing the effective cost per kilogram of the active intermediate. These factors collectively contribute to a more resilient and cost-effective supply chain capable of meeting the demands of commercial scale-up of complex pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The primary economic driver of this new process is the substitution of expensive proprietary coupling agents with inexpensive, bulk-available triphosgene. By removing the need for EDCI and HOBt, the bill of materials is drastically simplified, and the associated costs of purchasing and storing these sensitive reagents are eliminated. Additionally, the reduction in post-treatment steps means less labor and fewer man-hours are required for purification, further driving down the operational expenditure. The high reaction efficiency ensures that raw material utilization is maximized, preventing the financial loss associated with low-yielding condensation reactions common in older methodologies.
- Enhanced Supply Chain Reliability: From a sourcing perspective, the raw materials for this route—R-mandelic acid, triphosgene, and 4-nitrophenylethylamine—are widely available from multiple global suppliers, reducing the risk of single-source bottlenecks. The robustness of the reaction conditions, which tolerate slight variations in temperature and stoichiometry without compromising quality, ensures consistent batch-to-batch performance. This reliability is crucial for reducing lead time for high-purity pharmaceutical intermediates, allowing manufacturers to maintain tighter delivery schedules and respond more agilely to market fluctuations in demand for the final API.
- Scalability and Environmental Compliance: The process is inherently designed for industrial scalability, utilizing standard unit operations such as stirring, heating, and crystallization that are easily transferable from pilot plant to multi-ton production scales. The significant reduction in wastewater generation, due to the avoidance of extensive acid/base washing cycles, aligns with increasingly strict environmental regulations and sustainability goals. This eco-friendly profile not only mitigates regulatory risk but also lowers the costs associated with effluent treatment, making the process viable for long-term commercial production in regions with stringent environmental compliance standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a clear picture of the process capabilities. Understanding these details is essential for technical teams evaluating the feasibility of adopting this route for their own manufacturing pipelines. The answers highlight the specific advantages in terms of purity, yield, and operational simplicity that distinguish this method from conventional alternatives.
Q: What are the advantages of using triphosgene over EDCI/HOBt for this synthesis?
A: The triphosgene method avoids expensive coupling reagents like EDCI and HOBt, significantly reducing raw material costs and simplifying the post-treatment process by eliminating complex washing steps required to remove urea byproducts.
Q: What purity levels can be achieved with this novel synthetic route?
A: The patented process consistently achieves a chemical purity of 99.73% with a high isolated yield of 91%, surpassing the purity levels of conventional boron-mediated methods which typically reach only 98.65%.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the method utilizes easily obtainable raw materials and operates under mild conditions (15-70°C), making it highly feasible for commercial scale-up with reduced three-waste generation compared to traditional condensation methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (R)-2-hydroxy-N-(4-nitrophenylethyl)-2-phenylacetamide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes for high-value pharmaceutical intermediates like the mirabegron precursor. Our technical team has extensively analyzed the triphosgene-mediated pathway and possesses the expertise to implement this technology with precision and safety. We boast extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent supply of material that meets stringent purity specifications. Our rigorous QC labs are equipped to verify the 99.73% purity benchmark and monitor for any trace impurities, guaranteeing that every batch delivered supports your downstream API synthesis without compromise.
We invite potential partners to engage with our technical procurement team to discuss how this advanced synthesis method can be integrated into your supply chain. By leveraging our manufacturing capabilities, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us to request specific COA data and route feasibility assessments, allowing you to make informed decisions that drive both quality and profitability in your pharmaceutical manufacturing operations.