Advanced Modular Synthesis of Indomethacin and Analogues for Commercial API Production
Introduction to Next-Generation Indomethacin Synthesis
The pharmaceutical industry constantly seeks more efficient pathways for producing non-steroidal anti-inflammatory drugs (NSAIDs), particularly indomethacin and its diverse analogues which show promise in tumor resistance reversal. Patent CN110981781A discloses a groundbreaking synthetic method that shifts the paradigm from de novo indole ring construction to late-stage functionalization. This technology addresses critical bottlenecks in medicinal chemistry by enabling the direct introduction of substituents at the C2, C3, and N1 positions of the indole scaffold. By leveraging palladium-catalyzed reactions and oxidative coupling strategies, this method offers a streamlined route that significantly reduces the number of synthetic steps required for structure-activity relationship (SAR) studies. For global procurement and R&D teams, this represents a pivotal advancement in accessing high-purity pharmaceutical intermediates with greater speed and flexibility.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of indomethacin has relied heavily on classical methodologies such as the Fischer indole synthesis, which necessitates the construction of the indole ring for every specific analogue desired. As detailed in prior art references like Merck's approaches, this often requires distinct and sometimes obscure starting materials that are not commercially available, forcing manufacturers to engage in multi-step preparation of precursors. Furthermore, transition metal-catalyzed indole syntheses, while effective, often suffer from substrate limitations and the need for difficult-to-prepare diene or propargyl starting materials. These conventional routes result in lengthy synthetic sequences, increased waste generation, and higher production costs, making rapid iteration for drug discovery prohibitively expensive and time-consuming for supply chain managers.
The Novel Approach
In stark contrast, the methodology described in CN110981781A introduces a modular three-step strategy that begins with a readily available indole core. This innovative route allows for the sequential and selective functionalization of the molecule without the need for protecting groups or guiding groups. The process initiates with a palladium-catalyzed reaction to introduce alkyl or aromatic rings at the C2 position, followed by the installation of a carboxylic acid fragment at the C3 position via oxidative coupling, and concludes with N1 acylation. This logical disconnection simplifies the synthesis of complex analogues, as structural changes can be made by simply swapping reagents in the final steps rather than redesigning the entire synthetic pathway. 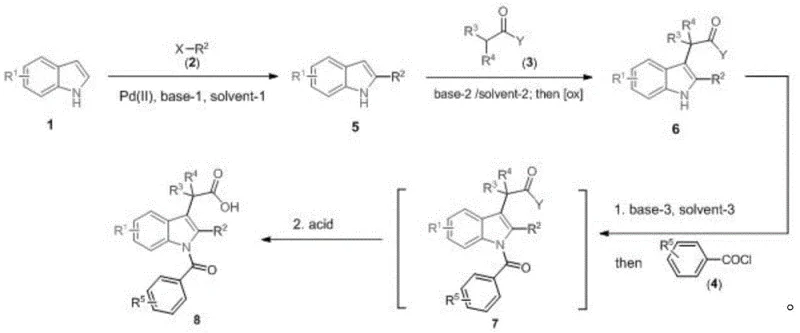
Mechanistic Insights into Pd-Catalyzed C2 Functionalization and Oxidative Coupling
The core of this technological breakthrough lies in the precise control of regioselectivity during the functionalization of the indole ring. The first step utilizes a Pd(II) catalyst, such as Pd(OAc)2 or PdCl2, in conjunction with a base like K2CO3 or Cs2CO3 in polar aprotic solvents like DMA or DMF. This catalytic cycle facilitates the direct coupling of halogenated compounds with the indole C2 position, a transformation that is traditionally challenging due to the inherent reactivity of the C3 position. Following this, the introduction of the carboxylic acid moiety at C3 is achieved through a sophisticated oxidative coupling mechanism. This involves the generation of an enolate using strong bases like LiHMDS at cryogenic temperatures (-78°C), followed by oxidation with ferric salts such as FeCl3. This sequence ensures high fidelity in placing the acetic acid side chain, which is critical for the biological activity of the final NSAID product.
Understanding the impurity profile is crucial for R&D directors focused on purity specifications. The method employs mild acidic workups, such as trifluoroacetic acid (TFA), to remove protecting groups like tert-butyl esters in the final stage, yielding the free carboxylic acid with high efficiency. The use of specific oxidants and the controlled addition of acyl chlorides in the final N-acylation step minimize the formation of regioisomers and over-acylated byproducts. For instance, the synthesis of indomethacin itself (Compound 8-1) proceeds through intermediate 6-1 and 7-1 with yields reaching up to 96% in the acylation step and 99% in the final deprotection, demonstrating exceptional chemoselectivity. 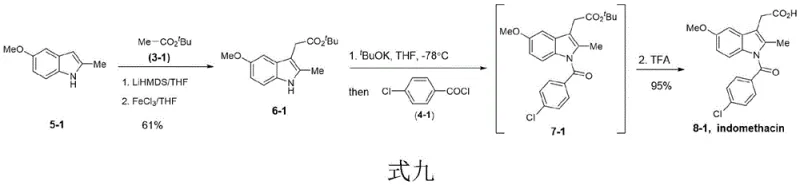
How to Synthesize Indomethacin Efficiently
The practical implementation of this synthesis route offers a clear advantage for process chemists aiming to establish robust manufacturing protocols. The procedure is designed to be operationally simple, utilizing standard laboratory equipment and commercially sourced reagents. The initial C2 functionalization can be performed at elevated temperatures (up to 150°C) with a wide range of palladium catalysts, providing flexibility in optimizing for cost or reaction rate. Subsequent steps require careful temperature control, particularly the lithiation and oxidation phases which are conducted at -78°C to prevent side reactions, but these conditions are standard in fine chemical manufacturing. Detailed standardized synthetic steps for the production of indomethacin and its analogues are provided below to guide technical teams in replication and scale-up.
- Perform Pd-catalyzed reaction to introduce alkyl or aromatic groups at the C2 position of the indole core.
- Execute oxidative coupling at the C3 position using strong bases like LiHMDS and oxidants such as FeCl3 to install the carboxylic acid fragment.
- Complete the synthesis by introducing the aroyl group at the N1 position using acyl chlorides under basic conditions, followed by acid hydrolysis.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this modular synthesis strategy translates directly into enhanced operational efficiency and risk mitigation. By decoupling the synthesis of the indole core from the installation of specific side chains, manufacturers can maintain a strategic stock of common intermediates, drastically reducing lead times for custom analogue production. This flexibility is invaluable in the fast-paced pharmaceutical sector where rapid response to clinical data is required. Furthermore, the reliance on commodity chemicals like indoles, palladium catalysts, and simple acyl chlorides ensures a stable supply chain that is less vulnerable to the shortages often associated with specialized, custom-synthesized starting materials used in older methods.
- Cost Reduction in Manufacturing: The elimination of multi-step indole ring construction for every new analogue results in significant cost savings. By shortening the synthetic route from potentially ten or more steps to just three functionalization steps, the consumption of raw materials, solvents, and energy is drastically reduced. Additionally, the high yields reported in the patent examples, such as the 96% yield in the acylation step, minimize material loss and waste disposal costs, contributing to a leaner and more cost-effective manufacturing process for high-purity pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The use of widely available starting materials and robust reaction conditions enhances the reliability of the supply chain. Unlike methods requiring unstable allenes or complex propargyl bromides, this process utilizes stable halides and indoles that are produced on a multi-ton scale globally. This ensures consistent availability and price stability, allowing procurement teams to negotiate better terms and secure long-term supply agreements for critical API intermediates without the fear of sudden raw material bottlenecks.
- Scalability and Environmental Compliance: The process is inherently scalable, moving seamlessly from gram-scale laboratory synthesis to multi-kilogram commercial production. The avoidance of hazardous reagents and the ability to use greener solvents in certain steps align with modern environmental compliance standards. The streamlined workflow reduces the overall solvent footprint and waste generation per kilogram of product, facilitating easier regulatory approval and supporting the company's sustainability goals in the production of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and claims within the patent documentation, providing a factual basis for decision-making. Understanding these details helps stakeholders evaluate the feasibility of integrating this method into their existing production pipelines for anti-inflammatory drugs and related therapeutic agents.
Q: How does this new synthesis method improve upon traditional Fischer Indole synthesis?
A: Traditional methods often require constructing the indole ring from scratch for each analogue, leading to long synthetic routes. This novel approach utilizes a modular strategy where the indole core is functionalized at C2, C3, and N1 positions sequentially, significantly shortening the route for analogue development.
Q: What are the key reaction conditions for the C3 carboxylation step?
A: The C3 carboxylation involves treating the C2-substituted indole with strong bases such as LiHMDS at low temperatures (-78°C), followed by oxidative coupling using ferric salts like FeCl3 to introduce the acetate fragment efficiently.
Q: Is this process suitable for large-scale manufacturing of NSAID intermediates?
A: Yes, the process utilizes commercially available indole starting materials and standard transition metal catalysts. The elimination of complex ring-building steps and the use of robust reaction conditions make it highly amenable to commercial scale-up and cost reduction in API manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Indomethacin Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this advanced synthesis technology in the landscape of NSAID production. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications required by global regulatory bodies, guaranteeing that every batch of indomethacin or its analogues meets the highest quality standards for pharmaceutical applications.
We invite you to collaborate with us to leverage this efficient synthetic route for your next project. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and target specifications. Please contact our technical procurement team today to request specific COA data and comprehensive route feasibility assessments, and let us help you optimize your supply chain for the next generation of anti-inflammatory therapeutics.