Revolutionizing Sartan Production: A Scalable One-Pot Route for Candesartan Cilexetil
Introduction to Advanced Sartan Synthesis
The pharmaceutical landscape for angiotensin II receptor blockers (ARBs) is constantly evolving, driven by the need for more efficient and environmentally sustainable manufacturing processes. Patent CN107089972B, published in May 2021, introduces a groundbreaking preparation method for candesartan cilexetil, a widely prescribed prodrug for hypertension. This technology represents a significant leap forward by consolidating multiple synthetic steps into a streamlined one-pot procedure, directly addressing the industry's demand for high-purity intermediates and reduced operational complexity. The core innovation lies in the seamless integration of alkylation, hydrolysis, and esterification reactions, bypassing the cumbersome protection and deprotection sequences that have historically plagued sartan synthesis. By leveraging this novel approach, manufacturers can achieve superior control over the impurity profile while drastically cutting down on processing time and solvent usage.
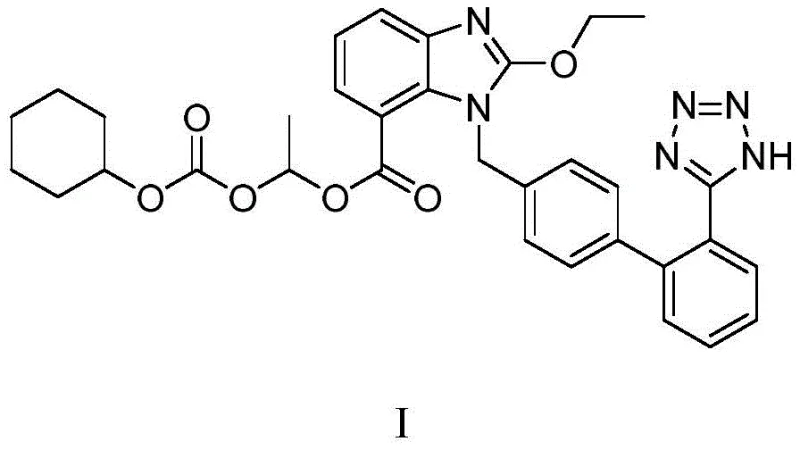
For R&D directors and process chemists, the structural integrity of the final active pharmaceutical ingredient (API) is paramount. The molecule, chemically defined as 2-ethoxy-1-{[2'-(1H-tetrazol-5-yl)(1,1'-biphenyl)-4-yl]methyl}benzimidazole-7-carboxylic acid-1-{[(cyclohexyloxy)carbonyl]oxy}ethyl ester, requires precise construction of the biphenyl-tetrazole axis. Traditional methods often struggle with the regioselectivity of the benzimidazole alkylation and the stability of the tetrazole ring under harsh conditions. This patent offers a robust solution that ensures the correct N-1 alkylation of the benzimidazole core while preserving the sensitive cyanide precursor until the final cyclization step. The result is a process that not only meets stringent regulatory standards for impurity limits but also provides a reliable pathway for consistent batch-to-batch quality in commercial production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of candesartan cilexetil has been burdened by inefficient multi-step sequences that rely heavily on protecting group chemistry. As illustrated in earlier literature such as EP459136, one common route involves the use of a triphenylmethyl (trityl) group to protect the tetrazole nitrogen during the alkylation phase. While effective for preventing side reactions, the removal of this bulky protecting group necessitates the use of strong acids, which can lead to the degradation of the sensitive benzimidazole core and the formation of difficult-to-remove acidic impurities. Furthermore, the introduction and subsequent removal of the trityl group add two entire synthetic steps, increasing the overall cycle time and consuming additional reagents and solvents without adding value to the final product structure.
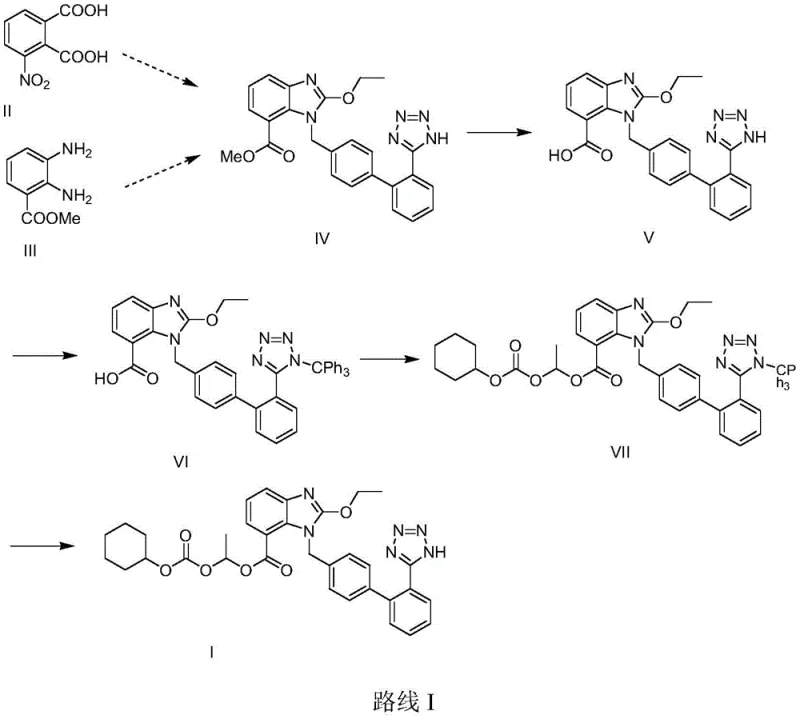
Another improved route, documented in CN1204125C, attempted to mitigate these issues by employing a tert-butoxy methyl aminobenzoate starting material. Although this method successfully omitted the N-protection and deprotection steps associated with the trityl group, it still suffered from redundancy in the synthetic sequence. The reliance on specific starting materials that require pre-functionalization means that the supply chain is dependent on niche intermediates rather than commodity chemicals. Additionally, the separation and purification of intermediates between each step in these conventional routes generate substantial amounts of solid and liquid waste, creating significant environmental compliance challenges and inflating the cost of goods sold (COGS) for the final API.
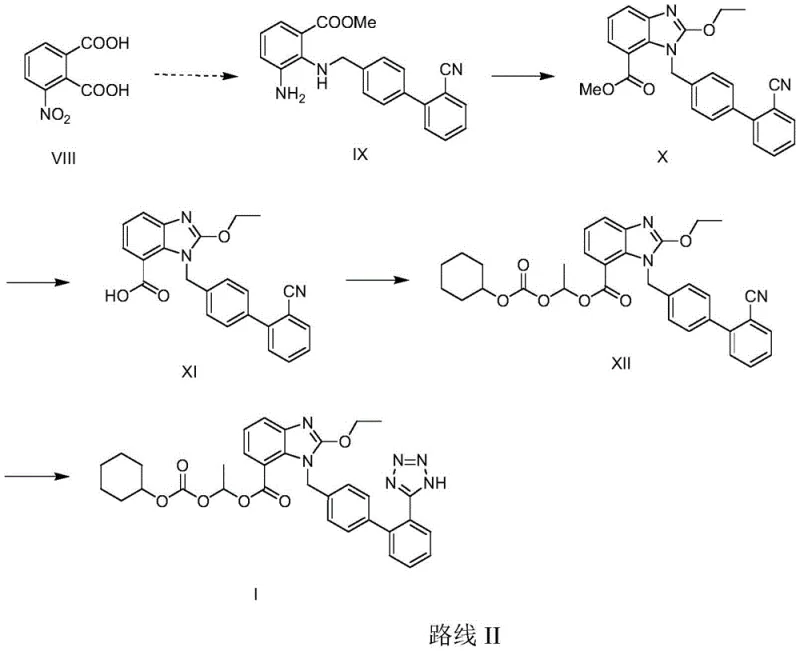
The Novel Approach
In stark contrast to these legacy methods, the process disclosed in CN107089972B utilizes a convergent strategy that maximizes atom economy and operational efficiency. The novel route begins with the direct alkylation of 2-ethoxybenzimidazole-7-alkyl formate (Compound XIII) with 4-bromomethyl-2'-cyanobiphenyl (Compound XIV). Instead of isolating the alkylated intermediate, the reaction mixture is immediately subjected to basic hydrolysis to reveal the carboxylic acid, followed by in-situ esterification with 1-halogenated ethyl cyclohexyl carbonate. This telescoping of three distinct chemical transformations into a single reactor vessel eliminates the need for intermediate isolation, filtration, and drying, which are typically the most time-consuming and yield-losing operations in batch processing.
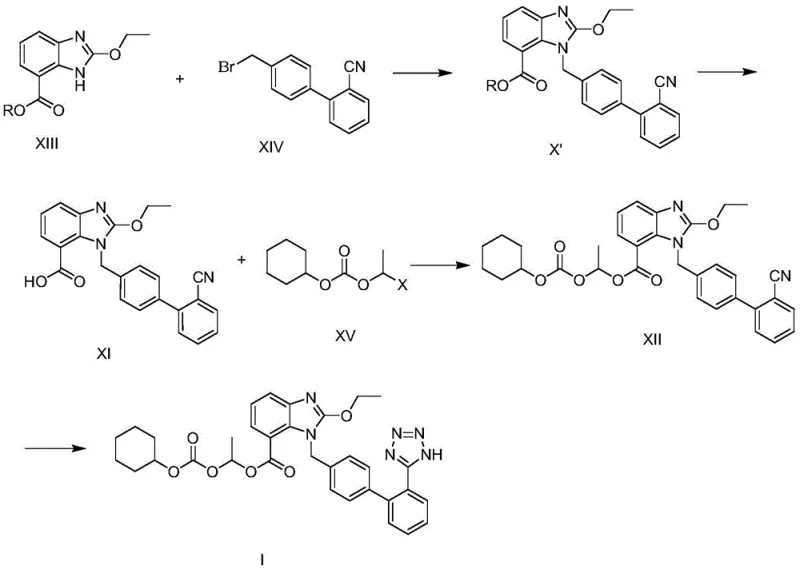
The strategic advantage of this one-pot methodology extends beyond mere time savings; it fundamentally alters the impurity landscape of the synthesis. By avoiding the isolation of the free carboxylic acid intermediate, the process minimizes exposure to potential decarboxylation or other degradation pathways that can occur during work-up. Furthermore, the final tetrazole formation is performed on the fully assembled carbonate ester intermediate, ensuring that the sensitive prodrug moiety is installed only after the core scaffold is securely established. This sequence allows for the use of robust Lewis acid catalysts like zinc chloride or tributyltin chloride to drive the [2+3] cycloaddition of the nitrile with sodium azide, resulting in high yields of the final API with exceptional purity profiles suitable for direct crystallization.
Mechanistic Insights into One-Pot Telescoped Alkylation and Esterification
The success of this synthesis hinges on the careful orchestration of nucleophilic substitution and acyl transfer reactions within a compatible solvent system. In the initial alkylation phase, the benzimidazole nitrogen acts as a nucleophile, attacking the benzylic bromide of the biphenyl component. The choice of base, preferably potassium carbonate (K2CO3), is critical here; it is strong enough to deprotonate the benzimidazole (pKa ~12-13 in DMSO) to facilitate the SN2 reaction but mild enough to prevent the hydrolysis of the starting ester or the nitrile group prematurely. The reaction is typically conducted in polar aprotic solvents like acetone or DMF at temperatures ranging from 50°C to 60°C, providing the necessary activation energy for the alkylation while maintaining the stability of the reactants.
Following the completion of alkylation, the addition of a stronger base such as potassium hydroxide (KOH) triggers the saponification of the methyl or ethyl ester at the 7-position of the benzimidazole ring. This hydrolysis proceeds rapidly under the existing thermal conditions to generate the carboxylate salt. Without isolating this salt, the introduction of 1-chloroethyl cyclohexyl carbonate initiates the esterification step. This reagent acts as an electrophilic source of the [(cyclohexyloxy)carbonyl]oxy]ethyl group, reacting with the in-situ generated carboxylate to form the crucial carbonate ester linkage. The compatibility of these sequential steps in one pot demonstrates a sophisticated understanding of chemoselectivity, ensuring that the base-mediated hydrolysis does not interfere with the subsequent alkylation of the carboxylic acid.
Impurity control is inherently built into this mechanistic design. In traditional routes, the isolation of the free acid intermediate often leads to dimerization or oligomerization impurities. By keeping the acid in its salt form and immediately converting it to the ester, the concentration of the reactive free acid species is kept negligible, effectively suppressing these side reactions. Additionally, the final tetrazole formation utilizes sodium azide and a Lewis acid catalyst at elevated temperatures (138-142°C). The use of Lewis acids like ZnCl2 coordinates with the nitrile nitrogen, increasing its electrophilicity and facilitating the attack by the azide ion. This catalytic cycle ensures complete conversion of the nitrile to the tetrazole ring, minimizing the risk of residual genotoxic nitrile impurities in the final drug substance, a critical quality attribute for regulatory approval.
How to Synthesize Candesartan Cilexetil Efficiently
The implementation of this patented technology requires precise control over reaction parameters to maximize the benefits of the one-pot design. The process begins by charging the reactor with the benzimidazole starting material, the biphenyl bromide, and a carbonate base in a solvent like acetone. Once the alkylation is confirmed complete via TLC or HPLC, the hydrolysis agent is added directly to the same mixture. After saponification, the carbonate esterifying agent is introduced, and the temperature is adjusted to drive the esterification to completion. The resulting intermediate is then isolated via simple filtration and crystallization, bypassing complex extraction workflows. For the final step, the intermediate is reacted with sodium azide and a Lewis acid in xylene or DMF to close the tetrazole ring, followed by a controlled acidic workup to precipitate the pure product.
- Perform alkylation of 2-ethoxybenzimidazole-7-alkyl formate with 4-bromomethyl-2'-cyanobiphenyl under alkaline conditions.
- Without isolation, proceed to hydrolyze the ester group and react with 1-halogenated ethyl cyclohexyl carbonate in the same pot.
- Convert the cyano group of the resulting intermediate into a tetrazole ring using sodium azide and a Lewis acid catalyst.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this synthesis route offers transformative economic benefits driven by process intensification. The consolidation of alkylation, hydrolysis, and esterification into a single vessel dramatically reduces the number of unit operations required per batch. This reduction translates directly into lower capital expenditure requirements for equipment, as fewer reactors and dryers are needed to achieve the same output volume. Furthermore, the elimination of intermediate isolation steps significantly cuts down on solvent consumption and the energy costs associated with solvent recovery and distillation. For procurement managers, this means a substantial reduction in the variable costs of manufacturing, allowing for more competitive pricing strategies in the generic pharmaceutical market without compromising margin.
- Cost Reduction in Manufacturing: The economic impact of removing protecting group chemistry cannot be overstated. Traditional routes require the purchase of expensive trityl chlorides or specialized protected starting materials, along with the strong acids needed for their removal. By utilizing commodity chemicals like 4-bromomethyl-2'-cyanobiphenyl and simple alkyl carbonates, the raw material bill of materials (BOM) is significantly optimized. Additionally, the one-pot nature of the reaction minimizes product loss during transfer and purification stages, leading to higher overall yields and better mass balance efficiency. This lean manufacturing approach ensures that every kilogram of raw material contributes more effectively to the final output, driving down the cost per kilogram of the active ingredient.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of widely available starting materials. The key building blocks, such as 2-ethoxybenzimidazole derivatives and cyanobiphenyl bromides, are produced by multiple global suppliers, reducing the risk of single-source bottlenecks. The simplified process flow also shortens the manufacturing lead time, enabling faster response to market demand fluctuations. With fewer steps comes fewer opportunities for batch failures or deviations, resulting in a more predictable and reliable production schedule. This stability is crucial for maintaining continuous supply to downstream formulation partners and avoiding costly stock-outs in the distribution network.
- Scalability and Environmental Compliance: Environmental sustainability is increasingly a key performance indicator for chemical manufacturers. This process generates significantly less waste compared to conventional methods, primarily due to the avoidance of extraction and washing steps between reactions. The post-treatment involves simple filtration to remove inorganic salts, followed by crystallization, which produces a cleaner mother liquor that is easier to treat or recycle. The reduction in wastewater volume and the minimization of hazardous acidic waste from deprotection steps align with strict environmental regulations. This green chemistry profile not only lowers waste disposal costs but also future-proofs the manufacturing site against tightening environmental legislation, ensuring long-term operational viability.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis route. These insights are derived directly from the experimental data and process descriptions provided in the patent documentation, offering clarity on how this technology can be integrated into existing manufacturing frameworks. Understanding these details is essential for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the strategic value of this supply chain optimization.
Q: How does the one-pot method improve purity compared to traditional routes?
A: By eliminating the isolation of intermediates and avoiding the use of triphenylmethyl protecting groups which require harsh acidic deprotection, the new route significantly reduces the formation of acid-sensitive impurities and simplifies the purification profile.
Q: What are the key cost drivers reduced in this synthesis?
A: The process eliminates expensive protecting group reagents and reduces solvent consumption by combining three reaction steps (alkylation, hydrolysis, esterification) into a single vessel, thereby lowering both material and waste disposal costs.
Q: Is this route suitable for large-scale commercial manufacturing?
A: Yes, the use of cheap, readily available raw materials like 4-bromomethyl-2'-cyanobiphenyl and the ability to recycle organic solvents makes this method highly scalable and economically viable for industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Candesartan Cilexetil Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and robust infrastructure. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the efficiencies promised by patent CN107089972B are fully realized at an industrial scale. We maintain stringent purity specifications through our rigorous QC labs, utilizing advanced analytical techniques to monitor critical quality attributes such as residual nitriles and enantiomeric excess. Our commitment to quality assurance guarantees that every batch of candesartan cilexetil intermediate meets the highest global regulatory standards, providing peace of mind for your regulatory filings.
We invite you to collaborate with us to optimize your supply chain for this critical antihypertensive agent. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this one-pot technology can improve your bottom line. We encourage you to contact us to request specific COA data and route feasibility assessments, allowing you to make informed decisions based on hard data rather than theoretical projections. Let us help you secure a competitive advantage in the sartan market through superior process chemistry and reliable supply.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →