Advanced Lenvatinib Manufacturing: Overcoming Genotoxic Impurities for Commercial Scale-up
The pharmaceutical industry continuously seeks robust synthetic pathways for multi-target tyrosine kinase inhibitors, particularly for oncology applications where purity standards are paramount. Patent CN113372270A introduces a significant advancement in the preparation of Lenvatinib, a critical medication approved for treating differentiated thyroid cancer. This technical disclosure addresses long-standing challenges regarding genotoxic impurity control and process scalability that have plagued earlier manufacturing methods. By re-engineering the urea formation step through an in-situ isocyanate strategy, the inventors have established a route that not only enhances chemical purity but also streamlines the downstream processing requirements. For global procurement teams and R&D directors, understanding this shift from traditional coupling methods to this optimized protocol is essential for securing a reliable Lenvatinib supplier capable of meeting stringent regulatory specifications. The structural integrity of the final mesylate salt, as depicted below, relies heavily on the precision of the preceding free base synthesis.
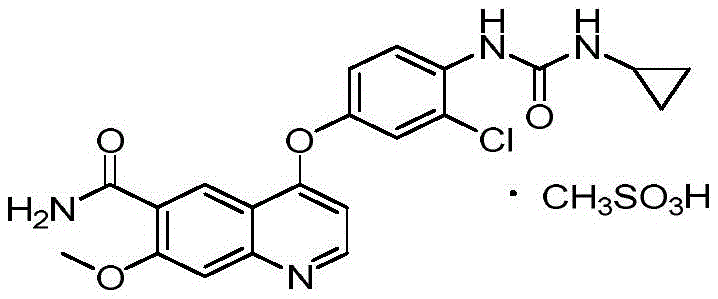
The evolution of Lenvatinib synthesis has been marked by iterative improvements aimed at resolving specific chemical bottlenecks. Early disclosures, such as those found in patent CN1878751A, described a linear route involving the acylation of 3-chloro-4-aminophenol followed by coupling with cyclopropylamine. While conceptually straightforward, this conventional approach suffers from a critical flaw: the persistence of genotoxic intermediates. Specifically, the unreacted aniline derivatives are notoriously difficult to purge from the final crystal lattice using standard recrystallization techniques. Furthermore, alternative routes attempting to couple the quinoline core later in the sequence often encounter yield losses due to the steric hindrance of the bulky substituents. These limitations necessitate extensive chromatographic purification, which is economically unsustainable for commercial scale-up of complex kinase inhibitors. The inability to consistently drive the reaction to completion results in batch-to-batch variability that poses significant risks to supply chain continuity.
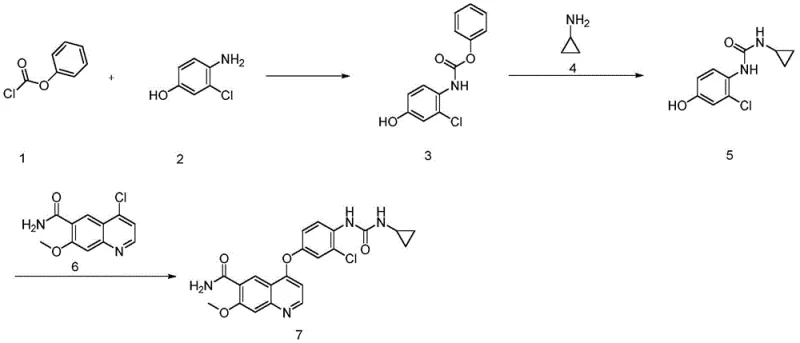
In stark contrast, the novel approach detailed in the subject patent fundamentally alters the reaction mechanism to bypass these pitfalls. Instead of relying on the direct coupling of unstable carbamates, the new methodology generates the reactive isocyanate species in situ. This is achieved by reacting cyclopropylamine with triphosgene under carefully controlled alkaline conditions. The immediate consumption of the generated isocyanate by the quinoline-phenol intermediate ensures that the concentration of reactive species remains low, thereby minimizing polymerization or hydrolysis side reactions. This strategic modification allows for a much tighter control over the impurity profile, specifically targeting the residual levels of the genotoxic starting materials. By shifting the equilibrium towards the product through stoichiometric precision, the process achieves a level of cleanliness that traditional methods cannot match without excessive waste generation. This represents a paradigm shift in cost reduction in API manufacturing, as it reduces the reliance on expensive scavenging resins or multiple recrystallization cycles.
Mechanistic Insights into In-situ Isocyanate Formation and Coupling
The core innovation lies in the precise management of the isocyanate formation step, which serves as the rate-determining phase of the synthesis. In this mechanism, triphosgene acts as a safe solid substitute for phosgene, decomposing under basic conditions to release the reactive carbonyl species. The reaction is conducted in a biphasic solvent system, typically involving toluene or dichloromethane and water, which facilitates the separation of inorganic salts formed during the neutralization of the hydrochloride byproducts. Maintaining the temperature between -10°C and 10°C is critical; temperatures that are too high accelerate the decomposition of the isocyanate, while temperatures that are too low hinder the reaction kinetics. The addition of a base, such as potassium carbonate or triethylamine, in specific molar excesses ensures that the amine is fully deprotonated and available for nucleophilic attack. This delicate balance prevents the accumulation of free isocyanate, which could otherwise react with moisture to form unwanted urea byproducts. The result is a highly efficient transformation that maximizes atom economy while maintaining a safe operating envelope for plant personnel.
Impurity control is further enhanced by the stoichiometric relationship between the isocyanate precursor and the quinoline intermediate. The patent specifies using a slight excess of the amine component relative to the triphosgene, ensuring that all toxic phosgene equivalents are consumed before the coupling step begins. Subsequently, the quinoline-phenol intermediate is introduced dropwise, allowing it to react immediately with the freshly generated isocyanate. This 'feed-and-react' strategy prevents the buildup of unreacted aniline, which is the primary genotoxic concern in this chemical class. Analytical data from the examples indicates that this method consistently drives the residual levels of the starting aniline down to parts-per-million (ppm) ranges, well below the thresholds required by international pharmacopoeias. Such rigorous control over the impurity spectrum is vital for high-purity Lenvatinib production, ensuring that the final drug substance meets the strict safety profiles demanded by regulatory agencies worldwide.
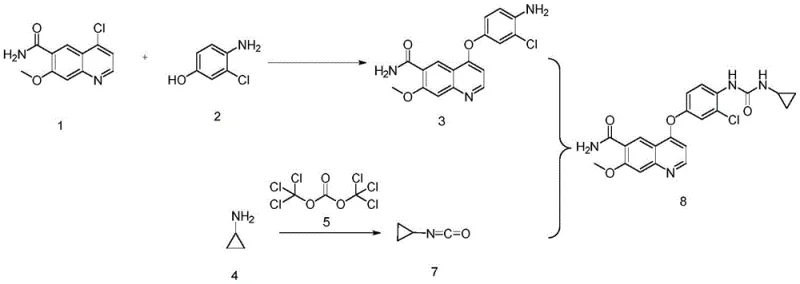
How to Synthesize Lenvatinib Efficiently
Implementing this synthesis route requires strict adherence to the operational parameters defined in the patent to ensure reproducibility and safety. The process is divided into three distinct stages: the formation of the ether-linked quinoline intermediate, the in-situ generation of the isocyanate, and the final coupling and isolation. Each stage demands precise temperature control and stoichiometric accuracy to maintain the integrity of the sensitive functional groups involved. The following guide outlines the critical workflow derived from the patent examples, serving as a foundational reference for process chemists aiming to replicate this high-efficiency pathway. Detailed standardized synthesis steps are provided in the guide below.
- Preparation of Key Intermediate: React 4-chloro-7-methoxyquinoline-6-carboxamide with 3-chloro-4-aminophenol under alkaline conditions at 65-75°C to form the ether linkage intermediate.
- In-situ Isocyanate Generation: React cyclopropylamine with triphosgene in a biphasic solvent system at low temperature (-10°C to 10°C) to generate cyclopropyl isocyanate without isolation.
- Final Coupling and Purification: Dropwise add the intermediate amine into the isocyanate solution, ensuring complete reaction to minimize genotoxic residue, followed by crystallization and drying.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this optimized synthetic route offers substantial benefits that extend beyond mere chemical elegance. For procurement managers, the primary value proposition lies in the simplification of the purification train. By minimizing the formation of hard-to-remove impurities at the source, the process eliminates the need for costly and time-consuming chromatographic separations. This directly translates to significant cost savings in manufacturing, as solvent consumption and waste disposal volumes are drastically reduced. Furthermore, the use of common industrial solvents like toluene and dichloromethane ensures that raw material sourcing remains stable and unaffected by niche supply constraints. The robustness of the reaction conditions also means that the process is less susceptible to minor fluctuations in utility supplies, enhancing overall operational reliability.
- Cost Reduction in Manufacturing: The elimination of extensive purification steps significantly lowers the operational expenditure associated with each production batch. By avoiding the use of expensive metal catalysts or specialized scavenging agents required in older routes, the overall cost of goods sold is optimized. The high crude purity achieved reduces the loss of valuable material during recrystallization, thereby improving the effective yield. This efficiency allows for more competitive pricing structures without compromising on quality standards, providing a distinct advantage in price-sensitive markets.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials and standard reagents mitigates the risk of supply disruptions. Unlike routes that depend on custom-synthesized building blocks with long lead times, this method utilizes commodity chemicals that can be sourced from multiple vendors. The robustness of the biphasic reaction system also allows for greater flexibility in manufacturing locations, enabling decentralized production strategies if necessary. This resilience is crucial for reducing lead time for high-purity pharmaceutical intermediates, ensuring that downstream formulation schedules are met without delay.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, utilizing reaction conditions that are easily transferable from pilot plants to large-scale reactors. The controlled generation of hazardous intermediates in situ minimizes the storage and handling risks associated with bulk isocyanates. Additionally, the reduced solvent usage and waste generation align with modern green chemistry principles, facilitating easier compliance with increasingly stringent environmental regulations. This sustainability aspect not only reduces regulatory burden but also enhances the corporate social responsibility profile of the supply chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Lenvatinib synthesis technology. These answers are derived directly from the experimental data and technical disclosures within the patent documentation, providing clarity on feasibility and performance. Understanding these details is key for stakeholders evaluating the potential for technology transfer or long-term supply partnerships.
Q: How does the new process control genotoxic impurities in Lenvatinib synthesis?
A: The process utilizes an in-situ generation of cyclopropyl isocyanate which reacts immediately with the amine intermediate. This ensures the genotoxic starting material (3-chloro-4-aminophenol derivative) is fully consumed, reducing residual levels to ppm ranges compared to traditional methods.
Q: What are the critical reaction conditions for the isocyanate formation step?
A: Critical control parameters include maintaining the reaction temperature between -10°C and 10°C and strictly controlling the molar equivalents of triphosgene (0.33-0.35 eq) and cyclopropylamine (1.2-1.8 eq) to prevent side reactions and ensure safety.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the method employs common solvents like toluene and dichloromethane and avoids unstable isolated intermediates. The simplified workup and high crude purity make it highly adaptable for multi-kilogram to ton-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Lenvatinib Supplier
The technical advancements described in patent CN113372270A represent the kind of innovation that drives the modern pharmaceutical supply chain forward. At NINGBO INNO PHARMCHEM, we recognize the critical importance of implementing such robust processes to ensure the consistent delivery of high-quality oncology APIs. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from development to market. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of Lenvatinib meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to leverage this advanced synthetic technology for your upcoming projects. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise can optimize your supply chain for Lenvatinib and other complex kinase inhibitors.