Advanced Synthesis of 2,5-Diazabicyclo[2.2.1]heptane Derivatives for Quinolone Antibiotics
Advanced Synthesis of 2,5-Diazabicyclo[2.2.1]heptane Derivatives for Quinolone Antibiotics
The pharmaceutical industry continuously seeks robust and scalable pathways for constructing complex heterocyclic scaffolds essential for next-generation therapeutics. Patent CN1025852C introduces a significant technological breakthrough in the preparation of 2,5-diazabicyclo[2.2.1]heptane derivatives, which serve as critical intermediates in the synthesis of potent quinolone antibiotics. This proprietary methodology offers a refined alternative to historical synthesis routes, leveraging readily available chiral pool materials like 4-hydroxyproline to establish the bicyclic core with high stereochemical fidelity. The innovation lies not merely in the final structure but in the strategic manipulation of functional groups to facilitate ring closure under milder conditions. ![General chemical structure of 2,5-diazabicyclo[2.2.1]heptane derivatives (Formula IA)](/insights/img/diazabicyclo-heptane-synthesis-pharma-supplier-20260315082555-01.webp) By utilizing sulfonyl protection strategies combined with selective reduction and nucleophilic substitution, this process addresses key pain points regarding safety, purity, and scalability that have long plagued the manufacturing of these nitrogen-rich heterocycles.
By utilizing sulfonyl protection strategies combined with selective reduction and nucleophilic substitution, this process addresses key pain points regarding safety, purity, and scalability that have long plagued the manufacturing of these nitrogen-rich heterocycles.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the 2,5-diazabicyclo[2.2.1]heptane skeleton relied heavily on methodologies described by Portoghese and others, which often involved harsh and hazardous reaction conditions. Traditional routes typically required the conversion of hydroxyproline into sulfonate esters followed by reaction with benzylamine, and subsequently, a rigorous reduction step utilizing hydriodic acid, red phosphorus, and acetic acid. These legacy processes present substantial operational risks due to the corrosive nature of hydriodic acid and the handling difficulties associated with red phosphorus. Furthermore, the multi-step nature of removing benzyl protecting groups often necessitates additional hydrogenation or strong acid treatments, which can compromise the integrity of sensitive functional groups elsewhere in the molecule. The accumulation of impurities from these aggressive reagents complicates downstream purification, leading to lower overall yields and increased waste generation, which is untenable for modern green chemistry standards.
The Novel Approach
In stark contrast, the method disclosed in CN1025852C employs a streamlined sequence that eliminates the need for hazardous phosphorus-based reductions. The novel approach initiates with the N-protection of hydroxyproline using sulfonyl chlorides, followed by a highly selective reduction of the carboxylic acid moiety to a primary alcohol using sodium borohydride activated by boron trifluoride etherate. This transformation is pivotal as it avoids the use of lithium aluminum hydride, offering better control over exothermicity and safety. Subsequent activation of the hydroxyl groups into leaving groups, such as tosylates or chlorides, sets the stage for an elegant intramolecular cyclization. By reacting these activated intermediates with simple alkylamines like methylamine in a sealed vessel, the bicyclic system is formed efficiently. This route not only simplifies the operational workflow but also enhances the safety profile of the manufacturing process, making it far more suitable for large-scale industrial application.
Mechanistic Insights into Stereoselective Cyclization and Reduction
The core of this synthetic strategy relies on precise stereochemical control derived from the chiral starting material. When 4-hydroxy-L-proline is employed, the resulting intermediates maintain specific configurations that dictate the final stereochemistry of the bicyclic product. The reduction of the carboxylic acid to the hydroxymethyl group using the NaBH4-BF3 complex proceeds with retention of configuration at the alpha-carbon, preserving the chiral information encoded in the proline ring. This is crucial because any epimerization at this stage would lead to diastereomeric impurities that are notoriously difficult to separate. The subsequent conversion of the hydroxyl groups into sulfonates or halides activates the molecule for nucleophilic attack without disturbing the stereocenters. 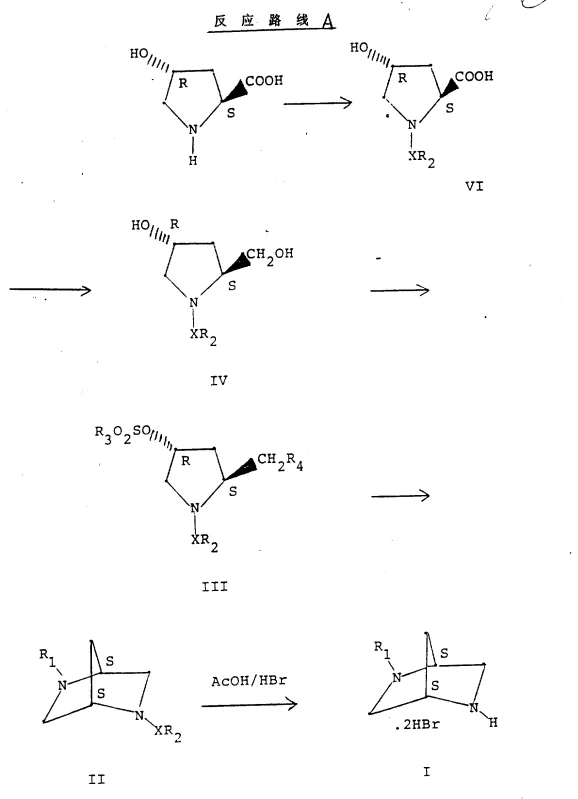 The cyclization step itself is an intramolecular SN2 reaction where the nitrogen atom attacks the activated carbon, displacing the leaving group. This displacement typically results in an inversion of configuration at the reaction center, a predictable outcome that allows chemists to design the synthesis backwards from the desired target geometry. The use of methylamine as the nucleophile introduces the second nitrogen atom required for the diazabicyclic framework, completing the core structure in a single, high-yielding step.
The cyclization step itself is an intramolecular SN2 reaction where the nitrogen atom attacks the activated carbon, displacing the leaving group. This displacement typically results in an inversion of configuration at the reaction center, a predictable outcome that allows chemists to design the synthesis backwards from the desired target geometry. The use of methylamine as the nucleophile introduces the second nitrogen atom required for the diazabicyclic framework, completing the core structure in a single, high-yielding step.
Impurity control is inherently built into this mechanism through the crystallinity of the intermediates. Many of the sulfonated intermediates, such as the bis-tosylates or chloro-tosylates described in the examples, are crystalline solids that can be purified by recrystallization before the final cyclization. This "purge point" strategy ensures that any unreacted starting material or side products generated during the activation steps are removed prior to the formation of the complex bicyclic system. Furthermore, the final deprotection step, which involves treating the N-sulfonyl bicyclic intermediate with hydrobromic acid in acetic acid, serves as a final cleanup. This acidic treatment cleaves the sulfonyl protecting groups and converts the free base into a stable hydrobromide salt, which often precipitates out of the reaction mixture in high purity. The ability to isolate the product as a salt significantly aids in meeting the stringent purity specifications required for pharmaceutical intermediates.
How to Synthesize 2,5-Diazabicyclo[2.2.1]heptane Efficiently
The synthesis of these valuable intermediates follows a logical progression from commodity amino acids to complex heterocycles, leveraging standard unit operations familiar to process chemists. The protocol begins with the protection of the amine and activation of the acid, followed by reduction and functional group interconversion to enable ring closure. Detailed standardized operating procedures for each transformation, including specific solvent choices, temperature profiles, and workup techniques, are essential for reproducibility. The following guide outlines the critical stages of this pathway, highlighting the specific reagents and conditions that maximize yield and stereochemical integrity. For the complete step-by-step experimental instructions and safety data, please refer to the structured guide below.
- Protect 4-hydroxy-L-proline with a sulfonyl group (e.g., tosyl) and reduce the carboxylic acid to a hydroxymethyl group using sodium borohydride and boron trifluoride.
- Activate the hydroxyl groups by converting them into leaving groups such as tosylates or chlorides using sulfonyl chlorides in pyridine.
- Perform intramolecular cyclization by reacting the activated intermediate with methylamine in a sealed vessel, followed by deprotection to yield the final diamine salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible benefits that extend beyond mere chemical elegance. The shift away from hazardous reagents like hydriodic acid and red phosphorus drastically reduces the regulatory burden and safety costs associated with raw material handling and waste disposal. By utilizing commodity chemicals such as tosyl chloride, sodium borohydride, and methylamine, the process insulates the supply chain from the volatility associated with specialized or controlled reagents. This reliance on widely available feedstocks ensures a more stable and continuous supply of the intermediate, reducing the risk of production stoppages due to raw material shortages. Additionally, the crystalline nature of key intermediates facilitates easier quality control and inventory management, as solid materials are generally more stable and easier to transport than oils or unstable solutions.
- Cost Reduction in Manufacturing: The elimination of expensive and dangerous reagents directly translates to lower raw material costs and reduced expenditure on safety infrastructure. The avoidance of benzyl protection groups, which require catalytic hydrogenation for removal, removes the need for precious metal catalysts and high-pressure hydrogenation equipment. This simplification of the equipment train lowers capital expenditure (CAPEX) and operational expenditure (OPEX). Furthermore, the high yields reported in the patent examples for each step minimize material loss, ensuring that the maximum amount of starting proline is converted into the valuable final product, thereby optimizing the cost per kilogram of the active intermediate.
- Enhanced Supply Chain Reliability: The use of 4-hydroxyproline as a chiral pool starter material leverages a robust global supply chain, as this amino acid is produced in large quantities for various industries. Unlike synthetic chiral auxiliaries that may have limited suppliers, hydroxyproline is available from multiple sources, mitigating single-source risk. The process conditions, which mostly operate at or near room temperature or moderate heating (e.g., 90°C in sealed vessels), are compatible with standard glass-lined or stainless steel reactors found in most multipurpose chemical plants. This compatibility means that the technology can be transferred to contract manufacturing organizations (CMOs) without requiring specialized or custom-built reactor setups, ensuring faster technology transfer and scale-up.
- Scalability and Environmental Compliance: From an environmental perspective, this route generates significantly less hazardous waste compared to traditional methods. The byproducts of the sulfonylation and reduction steps are generally inorganic salts and benign organic solvents that can be treated using standard wastewater protocols. The absence of heavy metals and phosphorus waste simplifies the environmental permitting process and reduces the long-term liability associated with hazardous waste storage. The scalability is further enhanced by the fact that the cyclization reaction, while requiring pressure vessels for gaseous methylamine, is a well-understood unit operation in the fine chemical industry. This ensures that the process can be scaled from pilot plant kilograms to multi-ton commercial production with predictable outcomes and minimal technical risk.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and claims within the patent documentation, providing a clear understanding of the process capabilities and limitations. Understanding these details is crucial for R&D teams evaluating the feasibility of this route for their specific pipeline projects and for procurement teams assessing the long-term viability of the supply source.
Q: What is the primary advantage of this synthesis route over conventional methods?
A: The primary advantage is the avoidance of hazardous reagents like hydriodic acid and red phosphorus used in older methods. This new route utilizes safer sulfonyl protection and mild reduction conditions, significantly improving process safety and environmental compliance while maintaining high stereochemical purity.
Q: Can this process produce both enantiomers of the diazabicyclic core?
A: Yes, the process is highly versatile. By selecting either 4-hydroxy-L-proline or 4-hydroxy-D-proline as the starting chiral pool material, manufacturers can selectively synthesize either the (S,S) or (R,R) stereoisomers of the 2,5-diazabicyclo[2.2.1]heptane scaffold, catering to specific antibiotic structural requirements.
Q: How is the stereochemistry controlled during the cyclization step?
A: Stereochemistry is controlled through an SN2-type intramolecular nucleophilic substitution. The configuration of the starting proline dictates the initial chirality, and the cyclization with methylamine proceeds with specific inversion at the reacting center, ensuring the formation of the desired exo- or endo- bicyclic geometry required for biological activity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2,5-Diazabicyclo[2.2.1]heptane Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving antibiotics. Our technical team has extensively analyzed the pathway described in CN1025852C and possesses the expertise to optimize this chemistry for commercial manufacturing. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and precision. Our state-of-the-art facilities are equipped with the necessary pressure reactors and corrosion-resistant lining to handle the specific conditions of this synthesis safely. With our rigorous QC labs and commitment to stringent purity specifications, we guarantee that every batch of 2,5-diazabicyclo[2.2.1]heptane derivative meets the highest international standards for pharmaceutical applications.
We invite you to collaborate with us to secure a stable and cost-effective supply of this essential intermediate. Our team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. By partnering with us, you gain access to a supply chain partner dedicated to driving efficiency and innovation in your antibiotic manufacturing processes, ensuring that you stay ahead in a competitive market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →