Scalable Synthesis of Diazabicycloheptane Intermediates for High-Purity Quinolone Antibiotics Production
The pharmaceutical landscape for quinolone antibiotics demands intermediates of exceptional stereochemical purity and structural integrity to ensure therapeutic efficacy and safety. Patent CN1047295A introduces a robust methodology for preparing 2,5-diazabicyclo[2.2.1]heptane derivatives, which serve as critical scaffolds in the synthesis of next-generation antibacterial agents. This technology leverages readily available chiral pool starting materials, specifically 4-hydroxy-L-proline or iso-4-hydroxy-D-proline, to construct the complex bicyclic nitrogen framework through a series of highly controlled transformations. By utilizing this patented approach, manufacturers can achieve superior control over stereocenters compared to older racemic synthesis methods, directly addressing the stringent regulatory requirements for chiral pharmaceutical ingredients. The process eliminates the need for difficult resolution steps later in the synthesis, thereby streamlining the overall production timeline and reducing material waste significantly.
Furthermore, the strategic design of this synthetic pathway allows for the introduction of various substituents at the nitrogen atoms, providing flexibility for derivative synthesis without compromising the core bicyclic stability. This adaptability is crucial for R&D teams exploring structure-activity relationships (SAR) in the development of novel quinolone variants. The use of stable sulfonate protecting groups ensures that the intermediate can be stored and transported without degradation, enhancing supply chain reliability for global pharmaceutical partners. As a reliable pharmaceutical intermediates supplier, understanding the nuances of such patented processes allows us to align our manufacturing capabilities with the precise needs of innovator companies seeking to secure their intellectual property and production continuity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
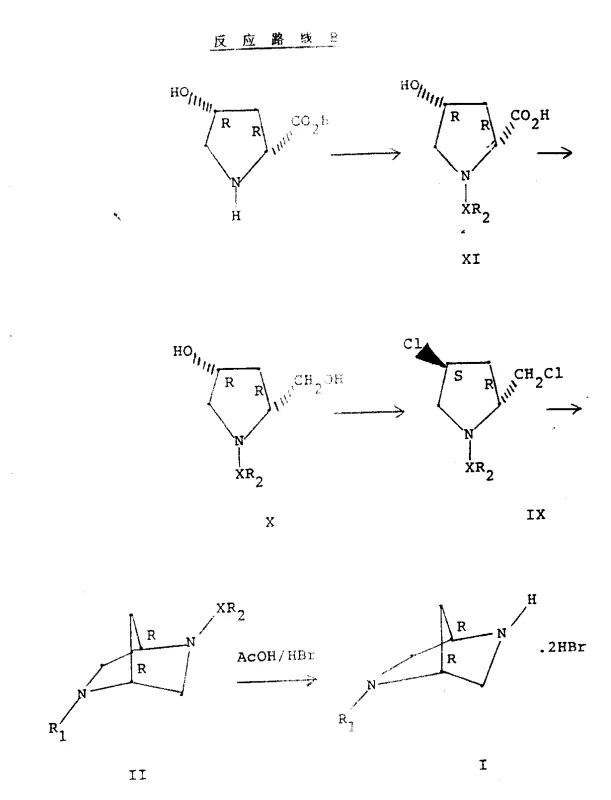
Historically, the synthesis of 2,5-diazabicyclo[2.2.1]heptane scaffolds relied heavily on methods described in earlier literature, such as those by Portoghese et al., which involved the conversion of hydroxyproline to tritosyl-hydroxy-L-prolinol followed by reaction with benzylamine and subsequent treatment with hydrogen iodide, phosphorus, and acetic acid. These conventional routes present significant safety and environmental challenges due to the use of corrosive hydrogen iodide and elemental phosphorus, which generate hazardous waste streams and require specialized containment equipment. Moreover, the multi-step conversion of dihydroiodide salts to the final methyl-substituted product often suffers from moderate yields and requires rigorous purification to remove residual iodine and phosphorus byproducts. Such impurities can be detrimental to downstream catalytic reactions in antibiotic synthesis, leading to catalyst poisoning and reduced overall process efficiency. The reliance on these harsh reagents also complicates regulatory approval processes, as residual heavy metals and halogens must be meticulously monitored and controlled to meet ICH guidelines.
The Novel Approach
In contrast, the methodology disclosed in CN1047295A offers a streamlined alternative that replaces hazardous reagents with safer, more manageable chemicals like tosyl chloride and sodium borohydride. This novel approach utilizes a direct cyclization strategy where activated pyrrolidine derivatives react with alkylamines in a sealed vessel, facilitating the formation of the bridged nitrogen system with high stereochemical fidelity. The process avoids the generation of toxic phosphine gases and minimizes the use of strong mineral acids, resulting in a cleaner reaction profile that simplifies work-up procedures and waste treatment. By employing commodity reagents and standard solvent systems such as methanol and tetrahydrofuran, this route supports cost reduction in pharmaceutical intermediates manufacturing while maintaining high product quality. The ability to access both S,S and R,R stereoisomers from distinct proline starting materials further enhances the utility of this method for producing enantiomerically pure antibiotics.
Mechanistic Insights into Stereoselective Cyclization and Protection
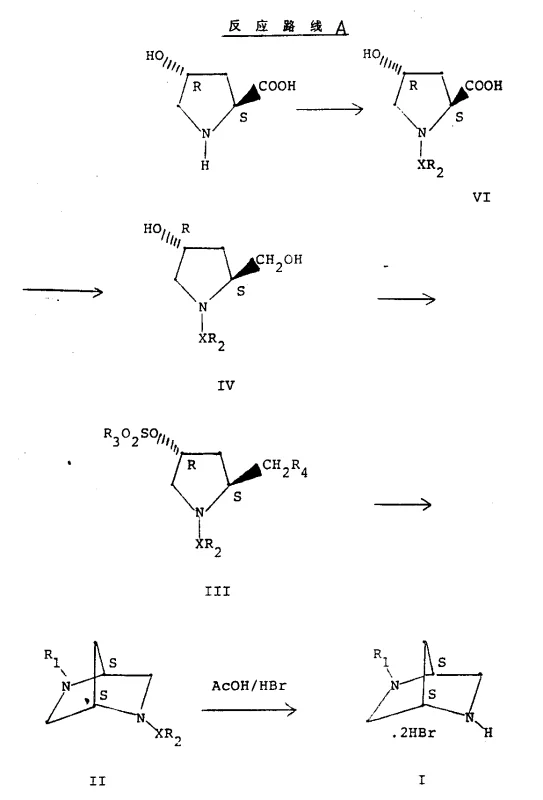
The core of this synthetic innovation lies in the precise manipulation of the proline ring to facilitate intramolecular nucleophilic substitution. The process begins with the protection of the secondary amine in 4-hydroxyproline using a sulfonyl group, typically a tosyl group, which serves dual purposes: it prevents unwanted side reactions at the nitrogen during subsequent steps and acts as a leaving group or stabilizing element during cyclization. Following protection, the carboxylic acid moiety is selectively reduced to a primary alcohol using a borane complex generated in situ from sodium borohydride and boron trifluoride etherate. This reduction step is critical as it sets the stage for the subsequent activation of the hydroxymethyl group, converting it into a good leaving group such as a tosylate or chloride. The stereochemistry at the C2 and C4 positions is preserved throughout these transformations, ensuring that the chirality of the starting proline is faithfully transferred to the final bicyclic product.
Subsequent activation of the hydroxyl groups creates a bis-electrophilic species capable of undergoing double nucleophilic attack by a primary amine, such as methylamine. This cyclization event constructs the 2,5-diazabicyclo[2.2.1]heptane core through a concerted mechanism that locks the conformational flexibility of the pyrrolidine ring into a rigid bicyclic structure. The rigidity of this scaffold is essential for the biological activity of the resulting quinolone antibiotics, as it positions the pharmacophore correctly for binding to bacterial DNA gyrase. Finally, the removal of the sulfonyl protecting group via acidic hydrolysis or reductive cleavage yields the free amine intermediate ready for coupling with quinolone precursors. This mechanistic pathway ensures high purity and minimizes the formation of regioisomers.
How to Synthesize 2,5-Diazabicyclo[2.2.1]heptane Efficiently
To implement this synthesis effectively, operators must adhere to strict control of reaction parameters, particularly temperature and stoichiometry during the cyclization phase. The detailed standardized synthesis steps involve initial tosylation in aqueous carbonate, followed by borohydride reduction in THF at controlled low temperatures to prevent exotherms. The subsequent activation and cyclization steps require sealed pressure vessels to maintain the concentration of gaseous methylamine, ensuring complete conversion to the bicyclic product. For comprehensive operational details, please refer to the structured guide below which outlines the specific molar ratios and safety protocols.
- Protect 4-hydroxy-L-proline with tosyl chloride in aqueous carbonate to form N-tosyl-4-hydroxy-L-proline.
- Reduce the carboxylic acid group using sodium borohydride and boron trifluoride etherate in THF to yield the corresponding alcohol.
- Activate hydroxyl groups via tosylation or chlorination, followed by cyclization with methylamine in a sealed vessel to form the diazabicyclic core.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the adoption of this synthetic route offers substantial strategic benefits regarding raw material availability and process safety. The reliance on bulk chemicals like tosyl chloride, sodium carbonate, and methylamine means that supply chains are less vulnerable to the fluctuations associated with specialized or scarce reagents. This stability translates into enhanced supply chain reliability, as manufacturers can source inputs from multiple global vendors without compromising on quality specifications. Furthermore, the elimination of hazardous phosphorus and hydrogen iodide reduces the regulatory burden and insurance costs associated with handling dangerous goods, contributing to significant cost savings in operational overhead. The high yields reported in the patent examples, often exceeding 80% to 90% for key steps, indicate a material-efficient process that minimizes waste disposal costs and maximizes output per batch.
- Cost Reduction in Manufacturing: The replacement of expensive and hazardous reagents with commodity chemicals drastically lowers the direct material cost per kilogram of the intermediate. Additionally, the simplified work-up procedures reduce solvent consumption and energy usage during distillation and drying phases. By avoiding complex purification steps required to remove phosphorus residues, the overall processing time is shortened, allowing for higher throughput in existing manufacturing facilities. These efficiencies collectively drive down the cost of goods sold, enabling more competitive pricing for the final antibiotic products in the global market.
- Enhanced Supply Chain Reliability: The use of stable intermediates and robust reaction conditions ensures consistent production schedules even in the face of minor operational variances. The ability to store protected intermediates without degradation provides a buffer against demand spikes, allowing for build-ahead strategies that secure inventory levels. This resilience is critical for maintaining continuous supply to downstream API manufacturers, preventing costly production stoppages due to intermediate shortages. The scalability of the process from laboratory to commercial tonnage further guarantees that supply can grow in tandem with market demand for quinolone antibiotics.
- Scalability and Environmental Compliance: The process generates aqueous waste streams that are easier to treat compared to the heavy metal and halogen-rich waste from conventional methods. This alignment with green chemistry principles facilitates easier permitting and compliance with increasingly strict environmental regulations. The reaction conditions, primarily operating between room temperature and 110°C, are compatible with standard glass-lined or stainless steel reactors, eliminating the need for exotic metallurgy. This compatibility accelerates technology transfer and scale-up, reducing the time to market for new drug formulations relying on this intermediate.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these diazabicyclic intermediates. The answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for industry professionals. Understanding these details helps stakeholders make informed decisions about integrating this technology into their existing supply chains.
Q: What is the primary advantage of this synthesis route over conventional methods?
A: This route avoids the use of hazardous hydrogen iodide and phosphorus, utilizing safer tosylation and borohydride reduction steps that offer better stereocontrol and higher yields.
Q: Can this process be scaled for commercial antibiotic production?
A: Yes, the reagents such as sodium carbonate, tosyl chloride, and methylamine are commodity chemicals, and the reaction conditions (room temperature to 110°C) are compatible with standard industrial reactors.
Q: What is the expected optical purity of the final intermediate?
A: Starting from enantiomerically pure 4-hydroxy-L-proline or iso-4-hydroxy-D-proline ensures the final diazabicycloheptane retains high optical purity, critical for quinolone efficacy.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2,5-Diazabicyclo[2.2.1]heptane Supplier
At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop to full-scale manufacturing. Our facility is equipped with stringent purity specifications and rigorous QC labs capable of verifying the stereochemical integrity and chemical purity of every batch according to pharmacopeial standards. We understand the critical nature of chiral intermediates in antibiotic synthesis and employ advanced analytical techniques to guarantee that our products meet the exacting requirements of global regulatory bodies. Our commitment to quality assurance ensures that you receive a consistent, high-performance intermediate that supports the efficacy and safety of your final pharmaceutical formulations.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain insights into potential efficiency gains and budget optimizations tailored to your production volume. We encourage you to contact us to obtain specific COA data and route feasibility assessments that demonstrate our capability to deliver this complex intermediate reliably. Let us partner with you to enhance your supply chain resilience and drive innovation in antibiotic development.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →