Advanced Synthesis of 3-Azabicyclo[3.1.0]hexane Derivatives for High-Purity Pharmaceutical Intermediates
Introduction to Novel 3-Azabicyclo[3.1.0]hexane Synthesis
The pharmaceutical industry continuously seeks robust and scalable pathways for constructing complex bicyclic scaffolds essential for next-generation antibiotics. Patent CN1265650A introduces a groundbreaking methodology for synthesizing special 3-azabicyclo[3.1.0]hexane derivatives featuring a double-protected amino group at the 6-position. This innovation addresses long-standing challenges in the preparation of gyrase inhibitors, specifically targeting the production of quinolone and 1,8-naphthyridinecarboxylic acid derivatives. Unlike traditional approaches that often struggle with stereocontrol and functional group compatibility during ring closure, this patented process integrates the amino functionality early in the synthetic sequence. By employing a dual-protection strategy using groups such as benzyl or allyl, the method ensures that the sensitive amine survives the rigorous conditions of cyclopropagation. This technical advancement not only enhances the purity profile of the resulting intermediates but also streamlines the overall manufacturing workflow, making it a highly attractive option for reliable pharmaceutical intermediate suppliers aiming to optimize their production lines for high-value antibacterial agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 6-amino-3-azabicyclo[3.1.0]hexane has been fraught with significant chemical and operational hurdles that impact both yield and cost efficiency. Conventional routes typically involve introducing the amino functionality only after the cyclopropylation step has been completed, necessitating complex functional group transformations on an already strained bicyclic system. This post-cyclization modification often leads to poor stereochemical outcomes, generating mixtures of endo and exo isomers that are difficult and expensive to separate. Furthermore, the use of monoprotected amines in prior art methods frequently results in instability during the ring-closing reactions, leading to polymerization or decomposition side products that contaminate the final API intermediate. These inefficiencies translate directly into higher manufacturing costs and extended lead times, as additional purification steps such as preparative chromatography become mandatory to meet stringent pharmaceutical purity specifications. The reliance on specific, often scarce, chiral auxiliaries in older methods further exacerbates supply chain vulnerabilities, creating bottlenecks that hinder the consistent availability of these critical building blocks for antibiotic production.
The Novel Approach
In stark contrast to legacy techniques, the novel approach detailed in the patent leverages a pre-functionalized strategy where the amino group, shielded by two robust protecting groups, is present in the molecule prior to the cyclopropylation event. This fundamental shift in synthetic logic allows for the direct conversion of chloroenamines or bicyclic nitriles into the desired 3-azabicyclo[3.1.0]hexane core with exceptional stereocontrol. Specifically, the reaction of chloroenamines with alkyl alcoholates generates N,O-acetal intermediates that subsequently undergo hydride reduction to exclusively yield the 6-endo-amine isomer. This high degree of stereoselectivity eliminates the need for costly isomer separation processes, drastically simplifying the downstream purification train. The versatility of the protecting groups, which can be benzyl, allyl, or other aryl-alkyl moieties, provides chemists with the flexibility to tailor the deprotection strategy to specific downstream requirements. By shifting the complexity to the earlier stages of synthesis where linear precursors are more manageable, this method achieves a level of process robustness that is ideal for commercial scale-up of complex pharmaceutical intermediates.
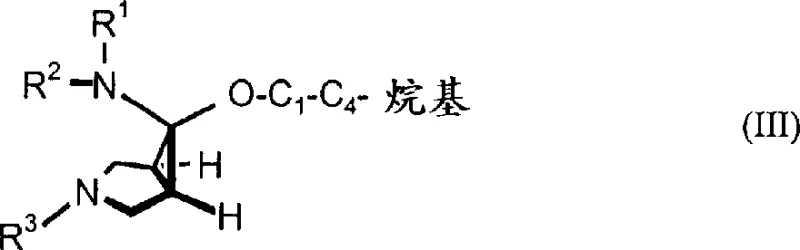
Mechanistic Insights into Stereoselective Cyclopropagation and Reduction
The core mechanistic advantage of this technology lies in the precise control exerted over the stereochemistry during the formation of the cyclopropane ring and the subsequent reduction of the N,O-acetal intermediate. When chloroenamines derived from 4-aminotetrahydropyridines react with alkyl alcoholates, the formation of the N,O-acetal proceeds through a transition state that inherently favors the endo-configuration due to steric and electronic factors imposed by the double-protected amine. This intermediate, characterized by a tertiary aminal unit, is then subjected to reduction using potent hydride conversion agents such as lithium aluminum hydride or diisobutylaluminum hydride. The hydride attack occurs with high facial selectivity, preserving the stereochemical integrity established in the previous step and ensuring that the resulting amine retains the desired 6-endo geometry. This mechanistic pathway is distinct from radical-based cyclizations which often suffer from racemization or loss of stereocontrol. Furthermore, the stability of the double-protected amine throughout this sequence prevents unwanted nucleophilic attacks on the electrophilic centers of the forming ring, thereby minimizing the formation of oligomeric impurities. Understanding this mechanism is crucial for R&D teams aiming to replicate these results, as parameters such as temperature and solvent choice during the hydride reduction phase play pivotal roles in maintaining the high diastereomeric excess observed in the patent examples.
Beyond the initial ring formation, the patent elucidates sophisticated mechanisms for the selective removal of protecting groups, which is vital for generating the final active pharmaceutical ingredient. For instance, benzyl groups can be cleanly removed via catalytic hydrogenation using palladium on carbon, a standard industrial process that leaves the sensitive bicyclic core intact. Alternatively, allyl groups can be cleaved using palladium catalysts in the presence of nucleophiles like barbituric acid derivatives, offering an orthogonal deprotection route that is compatible with other sensitive functionalities. The ability to selectively remove the N(3) substituent while retaining the 6-amino protection, or vice versa, allows for the modular assembly of diverse quinolone derivatives. This modularity is achieved through specific reactions such as treatment with vinyl chloroformate followed by acid hydrolysis, which targets specific carbamate linkages without affecting the rest of the molecular architecture. Such precise chemical control ensures that the impurity profile of the final drug substance remains within acceptable limits, reducing the burden on quality control laboratories and accelerating the regulatory approval timeline for new antibiotic candidates.
![Synthesis of quinolone derivatives using the novel 3-azabicyclo[3.1.0]hexane intermediate](/insights/img/3-azabicyclo-hexane-synthesis-pharma-supplier-20260308022206-07.webp)
How to Synthesize 6-Amino-3-azabicyclo[3.1.0]hexane Efficiently
Implementing this synthesis requires careful attention to reaction conditions, particularly during the preparation of the chloroenamine precursor and the subsequent cyclization steps. The process begins with the chlorination of substituted 4-aminotetrahydropyridines using N-chlorosuccinimide at low temperatures to prevent decomposition, followed by the addition of alkyl alcoholates to induce ring closure. The resulting N,O-acetal is then reduced under controlled conditions to yield the protected amine. While the general pathway is robust, optimizing parameters such as stoichiometry and addition rates is essential for maximizing yield and purity on a large scale. The detailed standardized synthesis steps see the guide below, which outlines the specific operational parameters required to achieve the high stereoselectivity reported in the patent literature. Adhering to these protocols ensures that manufacturers can consistently produce the 6-endo-amine isomer, which is the preferred configuration for minimizing neurotoxicity in the final quinolone antibiotic product.
- Preparation of chloroenamines from substituted 4-aminotetrahydropyridines via reaction with N-chlorosuccinimide at low temperatures.
- Cyclization and formation of N,O-acetals by reacting chloroenamines with alkyl alcoholates, ensuring high stereocontrol for the endo-isomer.
- Reductive removal of the alkoxy group using hydride conversion agents like lithium aluminum hydride to yield the final protected amine.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this patented methodology offers substantial strategic benefits that extend beyond mere chemical elegance. The elimination of complex chiral catalysts and the reliance on commodity chemicals like sodium methoxide and lithium aluminum hydride significantly reduces the raw material cost base, leading to cost reduction in pharmaceutical intermediate manufacturing. By avoiding the need for expensive enzymatic resolutions or precious metal-catalyzed asymmetric syntheses, the process becomes more resilient to fluctuations in the global market prices of specialized reagents. Furthermore, the high stereoselectivity inherent in the process means that fewer batch failures occur due to off-spec isomer ratios, thereby enhancing supply chain reliability and ensuring consistent delivery schedules to downstream API manufacturers. The robustness of the reaction conditions also implies that the process can be transferred between different manufacturing sites with minimal re-validation effort, providing supply chain heads with the flexibility to diversify their production footprint and mitigate geopolitical risks. Additionally, the use of standard unit operations such as crystallization and distillation for purification aligns well with existing infrastructure in most fine chemical plants, removing the need for capital-intensive equipment upgrades.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by eliminating the need for expensive chiral separation technologies and reducing the number of synthetic steps required to reach the key intermediate. Since the stereochemistry is controlled intrinsically by the reaction mechanism rather than by external chiral auxiliaries, the consumption of high-cost reagents is drastically minimized. Moreover, the high yields reported in the patent examples suggest that material throughput is maximized, reducing the waste disposal costs associated with low-efficiency processes. The ability to use common solvents and reagents further lowers the operational expenditure, making the overall cost of goods sold much more competitive compared to traditional routes that rely on proprietary catalysts.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials such as tetrahydropyridines and standard protecting group reagents ensures a stable supply of inputs, reducing the risk of production stoppages due to raw material shortages. The robustness of the chemistry allows for longer campaign runs without significant degradation in performance, which supports the continuous supply models required by large-scale antibiotic producers. Additionally, the process tolerance to minor variations in reaction conditions means that quality deviations are less frequent, ensuring that the supply of high-purity intermediates remains uninterrupted. This reliability is critical for maintaining the inventory levels necessary to support just-in-time manufacturing strategies in the pharmaceutical sector.
- Scalability and Environmental Compliance: The synthetic route is designed with scalability in mind, utilizing reaction conditions that are easily managed in large-scale reactors without requiring exotic pressure or temperature controls. The waste streams generated are primarily composed of standard organic solvents and inorganic salts, which can be treated using conventional wastewater treatment facilities, thereby simplifying environmental compliance. The avoidance of heavy metal catalysts in the main cyclization steps reduces the burden of metal residue testing and removal, streamlining the regulatory documentation required for drug master files. This environmental friendliness not only lowers compliance costs but also aligns with the increasing corporate sustainability goals of major pharmaceutical companies seeking green chemistry solutions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing a clear understanding of the process capabilities and limitations. Understanding these details is essential for technical teams evaluating the feasibility of adopting this route for their specific product portfolios. The information covers aspects ranging from stereochemical outcomes to deprotection strategies, ensuring a comprehensive overview of the technology's potential impact on production workflows.
Q: What is the primary advantage of the double-protection strategy in this synthesis?
A: The double-protection strategy allows the amino group to be present during the critical cyclopropylation step, preventing side reactions and enabling highly stereoselective formation of the 6-endo-amine isomer, which is crucial for low-neurotoxicity quinolones.
Q: How does this method improve supply chain reliability for antibiotic intermediates?
A: By utilizing robust starting materials like tetrahydropyridines and avoiding sensitive chiral catalysts, the process relies on standard chemical reagents and unit operations, significantly reducing the risk of supply disruptions and simplifying scale-up.
Q: Can the protecting groups be selectively removed?
A: Yes, the patent details multiple orthogonal deprotection methods, including hydrogenation for benzyl groups and specific cleavage protocols for allyl or vinyloxycarbonyl groups, allowing for flexible downstream modification.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Azabicyclo[3.1.0]hexane Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving antibiotics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in Patent CN1265650A can be successfully translated from the laboratory to the manufacturing plant. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to guarantee that every batch of 3-azabicyclo[3.1.0]hexane derivative meets the exacting standards required for pharmaceutical applications. Our commitment to technical excellence means that we can navigate the complexities of stereoselective synthesis and protecting group manipulation with precision, delivering intermediates that facilitate the efficient production of next-generation gyrase inhibitors.
We invite potential partners to engage with our technical procurement team to discuss how this advanced synthesis route can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain insights into the specific economic benefits of switching to this more efficient manufacturing method. We encourage you to contact us to obtain specific COA data and route feasibility assessments tailored to your project needs. Whether you require small quantities for clinical trials or metric tons for commercial launch, NINGBO INNO PHARMCHEM is positioned to be your trusted partner in delivering high-purity pharmaceutical intermediates with unmatched reliability and speed.