Industrial Scale-Up of 4,7-Dichloroquinoline: A High-Yield Route for Hydroxychloroquine Intermediates
Introduction to Patent CN103626699A
The pharmaceutical industry constantly seeks robust, scalable pathways for critical heterocyclic intermediates, particularly those serving as the backbone for antimalarial and autoimmune disease treatments. Patent CN103626699A, published in March 2014, introduces a highly efficient industrial preparation method for 4,7-dichloroquinoline, a pivotal precursor in the synthesis of hydroxychloroquine sulfate. This specific chemical entity is essential for treating discoid lupus erythematosus and systemic lupus erythematosus, representing a high-value niche within the fine chemical sector. The disclosed technology outlines a streamlined three-step sequence starting from ethyl 4-hydroxy-7-chloroquinoline-3-carboxylate, achieving a total yield of over 70% and a final product purity exceeding 99%. For R&D directors and supply chain managers, this patent represents a significant optimization over previous methodologies, offering a balance of high throughput and operational simplicity that is rare in complex heterocyclic synthesis.
The strategic value of this patent lies not just in the chemical transformation itself, but in its adaptability for large-scale manufacturing. By utilizing readily available raw materials and avoiding exotic catalysts, the process minimizes supply chain bottlenecks. The methodology employs standard unit operations such as reflux, filtration, and crystallization, which are easily replicated in multi-purpose chemical plants. Furthermore, the rigorous control of reaction parameters, such as maintaining specific pH levels during acidification and precise temperature ranges during decarboxylation, ensures consistent batch-to-batch quality. This level of process control is critical for pharmaceutical intermediates where impurity profiles can dictate the success of downstream API synthesis. As we delve deeper into the technical specifics, it becomes clear why this route is preferred for reliable pharmaceutical intermediate supplier networks aiming for cost-effective production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinoline derivatives has often been plagued by harsh reaction conditions, low atom economy, and difficult purification steps. Prior art, such as the methods described in background references like CN1847226A, focuses heavily on constructing the quinoline ring system from basic anilines and malonates. While effective for building the core structure, these upstream processes often leave manufacturers with intermediates that are challenging to functionalize further without significant degradation or yield loss. Traditional chlorination methods, for instance, might require excessive amounts of chlorinating agents or result in mixtures of regioisomers that are costly to separate. Additionally, older decarboxylation techniques often relied on solvent-free heating or copper catalysts, which could lead to charring, tar formation, and substantial losses in material throughput. These inefficiencies translate directly into higher production costs and extended lead times, creating friction for procurement managers seeking cost reduction in pharma manufacturing.
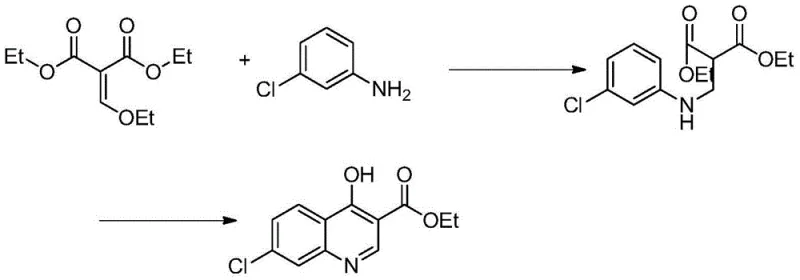
Moreover, the lack of a standardized, high-yield domestic report for converting the carboxylic acid ester directly to the dichloro derivative created a gap in the supply chain. Without a dedicated, optimized pathway, manufacturers were forced to cobble together generic literature procedures that were not validated for industrial tonnage. This often resulted in variable purity levels, necessitating multiple recrystallizations that further eroded overall yield. The reliance on transition metals or complex catalytic systems in alternative routes also introduced the risk of heavy metal contamination, requiring expensive scavenging steps to meet stringent regulatory limits for pharmaceutical ingredients. These cumulative drawbacks highlight the necessity for a dedicated, linear process that prioritizes both yield and purity from the outset.
The Novel Approach
The approach detailed in CN103626699A breaks away from these constraints by focusing on a linear, three-step refinement of the ethyl 4-hydroxy-7-chloroquinoline-3-carboxylate scaffold. Instead of attempting to build the ring and install the chlorine atoms simultaneously, this method decouples the transformations into distinct, controllable stages: hydrolysis, decarboxylation, and chlorination. This modularity allows for precise optimization of each step independently. For example, the hydrolysis step uses a mild 10% sodium hydroxide solution, avoiding the aggressive conditions that might degrade the sensitive quinoline nitrogen or the chloro substituent. The subsequent decarboxylation utilizes paraffin oil as a high-boiling heat transfer medium, ensuring uniform heating at 230-250°C without the localized hot spots that cause decomposition in solvent-free methods. Finally, the chlorination with phosphorus oxychloride is conducted in toluene, facilitating easy removal of byproducts and simplifying the workup.
This novel sequence effectively transforms a potentially messy multi-step synthesis into a predictable industrial workflow. By starting with a pre-functionalized ester, the process bypasses the need for difficult regioselective chlorinations on the quinoline ring, as the 7-chloro position is already established. The innovation lies in the efficient removal of the 3-carbethoxy group and the conversion of the 4-hydroxyl group to a chloride with minimal side reactions. The result is a process that is not only chemically elegant but also commercially viable, offering a clear path for commercial scale-up of complex pharmaceutical intermediates. The simplicity of the workup procedures, involving basic phase separations and crystallizations, means that the technology can be transferred to manufacturing sites with standard glass-lined or stainless steel reactors, enhancing supply chain reliability.
Mechanistic Insights into the Three-Step Transformation
The core of this technology relies on a classic yet meticulously optimized sequence of organic transformations. The first stage involves the saponification of the ethyl ester to the corresponding carboxylic acid. Using a 10% NaOH solution at a molar ratio of 1:1.5 to 1:2 relative to the substrate ensures complete conversion while minimizing excess base that could complicate neutralization. Heating the mixture to 90-100°C facilitates the nucleophilic attack of the hydroxide ion on the carbonyl carbon. Following hydrolysis, the reaction mixture is treated with activated carbon to remove colored impurities, a critical step for ensuring the high visual quality of the final API. The pH is then carefully adjusted to 3-4 using 10% hydrochloric acid. This specific pH range is chosen to precipitate the zwitterionic carboxylic acid form while keeping inorganic salts in solution, allowing for high-purity isolation via suction filtration with yields consistently above 90%.
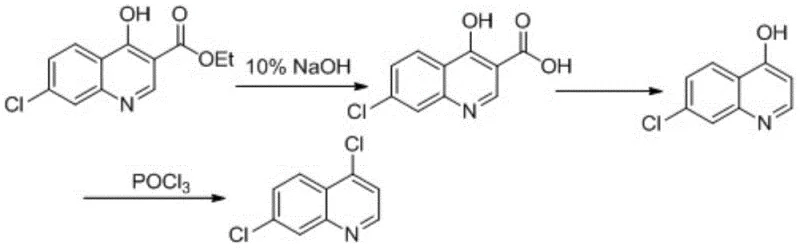
The second mechanistic phase is thermal decarboxylation, a reaction driven by entropy and the stability of the resulting aromatic system. By suspending the carboxylic acid intermediate in paraffin oil or light diesel and heating to 230-250°C, the molecule undergoes a concerted pericyclic-like transition state where the carboxyl group is expelled as carbon dioxide. The high boiling point of the paraffin medium is essential here, as it allows the reaction to proceed at the necessary activation energy without the solvent evaporating. This step is remarkably clean, with yields ranging from 90% to 100%, indicating that the quinoline nucleus remains intact under these thermal stresses. The final step is the chlorodehydroxylation using phosphorus oxychloride (POCl3). In the presence of toluene, POCl3 activates the 4-hydroxyl group, likely forming a phosphorylated intermediate that is subsequently displaced by a chloride ion. The reaction is maintained at reflux (90-115°C) to drive the equilibrium forward. The workup involves quenching in ice water to decompose excess POCl3, followed by careful pH adjustment to 7-8 to neutralize acidic byproducts, ensuring the final crystalline product achieves a purity of ≥99%.
How to Synthesize 4,7-Dichloroquinoline Efficiently
Implementing this synthesis requires strict adherence to the thermal and stoichiometric parameters defined in the patent to ensure safety and reproducibility. The process begins with the preparation of the acid intermediate, followed by the high-temperature decarboxylation which requires specialized heating equipment capable of sustaining 250°C safely. The final chlorination step demands careful handling of phosphorus oxychloride due to its corrosive nature and reactivity with water. Operators must ensure that the quenching into ice water is performed slowly to manage the exotherm and gas evolution effectively. The detailed standardized synthetic steps, including specific stirring rates, cooling profiles, and filtration techniques required to replicate the >70% total yield, are outlined below for technical teams preparing for pilot trials.
- Perform alkaline hydrolysis using 10% NaOH at 90-100°C, followed by acidification to pH 3-4 to isolate 4-hydroxy-7-chloroquinoline-3-carboxylic acid.
- Execute thermal decarboxylation in paraffin oil or light diesel at 230-250°C for 30-60 minutes to yield 4-hydroxy-7-chloroquinoline.
- Conduct chlorination using phosphorus oxychloride in toluene at reflux (90-115°C), followed by aqueous workup and recrystallization to achieve >99% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented process offers tangible strategic benefits beyond mere chemical yield. The primary advantage is the drastic simplification of the raw material portfolio. By relying on commodity chemicals such as sodium hydroxide, hydrochloric acid, paraffin oil, and phosphorus oxychloride, the process eliminates dependence on scarce or geographically constrained reagents. This ubiquity of inputs significantly enhances supply chain reliability, reducing the risk of production stoppages due to material shortages. Furthermore, the high yield at each individual step (>90% for the first two steps) means that less raw material is wasted per kilogram of finished product. This efficiency directly translates to cost reduction in pharma manufacturing, as the effective cost of goods sold (COGS) is lowered through better material utilization and reduced waste disposal fees.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts is a major financial driver. Traditional methods often require copper or palladium catalysts which are not only expensive to purchase but also costly to remove to meet ppm-level specifications. By avoiding these metals entirely, the process removes the need for expensive scavenging resins or complex extraction protocols. Additionally, the use of paraffin oil as a recyclable heat transfer medium reduces solvent consumption costs compared to methods requiring large volumes of volatile organic solvents for high-temperature reactions. The high purity of the crude product (>99% after simple recrystallization) minimizes the need for extensive chromatographic purification, which is notoriously expensive and difficult to scale. These factors combine to create a leaner, more cost-effective production model.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes to shorter cycle times and higher equipment turnover. Because the reactions are high-yielding and clean, the incidence of batch failures due to side reactions or impurity buildup is significantly reduced. This predictability allows for tighter production scheduling and more accurate delivery commitments to downstream API manufacturers. The process is also less sensitive to minor fluctuations in raw material quality, providing a buffer against supply variability. For a reliable pharmaceutical intermediate supplier, this consistency is key to maintaining long-term contracts with multinational pharmaceutical companies who demand uninterrupted supply for their global drug formulations.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, the process is designed for scalability. The unit operations—filtration, reflux, and crystallization—are standard in the fine chemical industry, meaning the technology can be scaled from 100 kgs to 100 MT annual commercial production without fundamental engineering changes. The waste stream is primarily aqueous salt solution and spent paraffin oil, both of which are easier to treat and dispose of compared to heavy metal-contaminated waste. The absence of halogenated solvents in the early steps and the containment of POCl3 in the final step within a closed reflux system align with modern green chemistry principles. This compliance reduces the regulatory burden and potential fines associated with hazardous waste management, further improving the overall economic viability of the project.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. They are derived from the specific operational parameters and benefits highlighted in patent CN103626699A. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this intermediate into their existing supply chains. The answers reflect the empirical data provided in the patent examples, ensuring accuracy for decision-making purposes.
Q: What is the overall yield and purity of the 4,7-dichloroquinoline produced via this method?
A: According to patent CN103626699A, the total yield of the product is greater than or equal to 70%, with individual step yields exceeding 90% for hydrolysis and decarboxylation, and over 80% for chlorination. The final product purity is consistently reported to be greater than or equal to 99%.
Q: Why is paraffin oil used in the decarboxylation step?
A: Paraffin oil or light diesel serves as a high-boiling solvent and heat transfer medium, allowing the reaction mixture to reach the necessary temperatures of 230-250°C required for efficient thermal decarboxylation without degrading the quinoline scaffold.
Q: How does this process address environmental concerns compared to traditional methods?
A: The process utilizes common reagents like sodium hydroxide and phosphorus oxychloride with straightforward workup procedures involving phase separation and recrystallization, resulting in less environmental pollution and simpler waste treatment protocols suitable for industrial compliance.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4,7-Dichloroquinoline Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the global pharmaceutical supply chain. Our technical team has extensively analyzed the pathway described in CN103626699A and possesses the expertise to execute this chemistry with precision. We understand that scaling a three-step sequence involving high-temperature decarboxylation and aggressive chlorination requires specialized reactor configurations and rigorous safety protocols. Our facility is equipped to handle these specific challenges, ensuring that every batch meets the stringent purity specifications required for hydroxychloroquine synthesis. With extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, we offer a partnership model that prioritizes both technical excellence and commercial reliability.
We invite procurement leaders and R&D directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific volume requirements. Our technical procurement team is ready to provide specific COA data and route feasibility assessments to demonstrate how our implementation of this patented process can optimize your supply chain. By choosing NINGBO INNO PHARMCHEM, you secure a partner dedicated to delivering high-purity 4,7-dichloroquinoline with the consistency and transparency needed to support your long-term drug development and commercialization goals.