Advanced Besifloxacin Manufacturing: A High-Yield Fluoro-Substitution Strategy for Global API Supply
Advanced Besifloxacin Manufacturing: A High-Yield Fluoro-Substitution Strategy for Global API Supply
The pharmaceutical industry continuously seeks robust synthetic pathways for critical anti-infective agents, particularly fluoroquinolones used in ophthalmic applications. Patent CN103044397A introduces a transformative methodology for synthesizing besifloxacin, a broad-spectrum antibiotic effective against bacterial conjunctivitis. This innovation addresses longstanding inefficiencies in traditional manufacturing by shifting the starting material strategy from chlorinated benzoic acids to fluorinated analogues. The core breakthrough lies in the utilization of 2,4,5-trifluorobenzoic acid, which offers superior reactivity and cost profiles compared to the conventional 5-chloro-2,4-difluorobenzoic acid. By deferring the introduction of the chlorine atom to the final synthetic stage, the process minimizes side reactions and maximizes overall yield. This technical evolution represents a significant leap forward for reliable pharmaceutical intermediates supplier networks aiming to optimize production scalability.
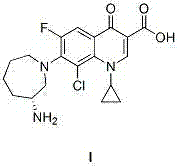
Besifloxacin, chemically known as (R)-7-(3-aminohexahydro-1H-azepin-1-yl)-8-chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-quinoline-3-carboxylic acid, demands precise stereochemical control. The structural complexity requires a synthesis that preserves chirality while ensuring high purity. The patent details a route that not only simplifies the operational steps but also enhances the stability of intermediates, facilitating easier storage and transportation. For procurement managers and supply chain heads, understanding these mechanistic shifts is vital for securing long-term cost reduction in pharmaceutical intermediates manufacturing. The transition to a fluoro-first strategy fundamentally alters the economic landscape of producing this high-value ophthalmic agent.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of besifloxacin has relied heavily on 5-chloro-2,4-difluorobenzoic acid as the primary building block. While chemically feasible, this traditional pathway suffers from inherent kinetic and thermodynamic disadvantages. The presence of the chlorine atom at the 5-position creates significant steric and electronic hurdles during nucleophilic substitution reactions. Chlorine is a poorer leaving group compared to fluorine in aromatic nucleophilic substitution contexts, necessitating harsher reaction conditions such as elevated temperatures and extended reaction times. These severe conditions often lead to the formation of undesirable by-products, complicating downstream purification and reducing the overall yield of the target intermediate. Furthermore, the reactivity of the chlorine atom can trigger premature side reactions, leading to a complex impurity profile that requires expensive chromatographic separation. For large-scale operations, these factors translate into higher energy consumption, increased solvent usage, and ultimately, a higher cost of goods sold (COGS).
The Novel Approach
The methodology disclosed in CN103044397A circumvents these bottlenecks by employing 2,4,5-trifluorobenzoic acid as the starting material. This strategic substitution leverages the higher reactivity of the fluorine atom, which acts as a superior leaving group during the critical condensation steps. The new route involves condensing the quinolone core intermediate (II) with L-aminocaprolactam to form intermediate (III), followed by a selective chlorination step using chlorosulfonic acid. By postponing the introduction of the chlorine atom until the final stage, the process effectively isolates the reactive halogenation event from the earlier sensitive condensation steps. This temporal separation of reactive functionalities drastically reduces the probability of side reactions, resulting in a cleaner reaction profile. The patent reports yields reaching up to 93% in the final chlorination step, a substantial improvement over conventional methods. This approach not only streamlines the workflow but also aligns perfectly with the goals of commercial scale-up of complex pharmaceutical intermediates.
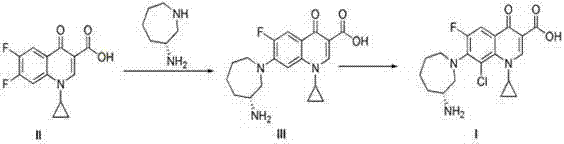
Mechanistic Insights into Late-Stage Chlorination and Fluoro-Displacement
The success of this novel synthesis hinges on the nuanced understanding of nucleophilic aromatic substitution (SnAr) mechanisms tailored for fluoroquinolone cores. In the condensation step, the piperazine-like nitrogen of the L-aminocaprolactam side chain attacks the C-7 position of the quinolone ring. The presence of the fluorine atom at this position, activated by the adjacent carbonyl groups, facilitates a smooth displacement. The use of bases such as triethylamine, potassium carbonate, or pyridine in acetonitrile solvent creates an optimal environment for this transformation. Acetonitrile, being a polar aprotic solvent, stabilizes the transition state without solvating the nucleophile too strongly, thereby enhancing its reactivity. The reaction proceeds under reflux conditions, typically around 10 hours, ensuring complete conversion while maintaining the integrity of the chiral center on the azepine ring. This preservation of stereochemistry is critical, as the (R)-enantiomer is the biologically active form required for therapeutic efficacy.
Following the formation of the key intermediate (III), the process employs a highly selective chlorination strategy. Chlorosulfonic acid serves as both the chlorinating agent and the acidic catalyst. The mechanism likely involves the activation of the aromatic ring by the strong acid, followed by electrophilic attack or a radical-mediated substitution at the C-8 position, displacing the remaining fluorine atom. Conducting this reaction in chloroform under controlled temperature gradients—starting from an ice bath and gradually warming to reflux—allows for precise control over the reaction kinetics. This careful thermal management prevents over-chlorination or degradation of the sensitive quinolone scaffold. The result is a high-purity product with minimal racemization, addressing a common pain point in chiral drug synthesis. The ability to maintain configuration constant from the L-lysine starting material through to the final API demonstrates a robust control over the stereogenic elements of the molecule.
How to Synthesize Besifloxacin Efficiently
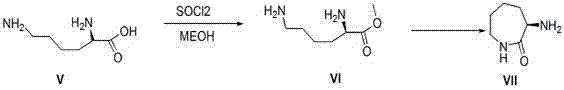
Implementing this synthesis requires precise adherence to the reaction parameters outlined in the patent to ensure reproducibility and safety. The process begins with the preparation of the chiral side chain, L-aminocaprolactam, derived from L-lysine methyl ester via cyclization. This intermediate is then coupled with the difluoro-quinolone core in acetonitrile. The subsequent chlorination step demands careful handling of chlorosulfonic acid due to its corrosive nature. Detailed standard operating procedures (SOPs) are essential for managing the exothermic nature of the chlorination and the workup phases involving neutralization and crystallization. For R&D teams looking to adopt this technology, the following guide outlines the critical operational milestones derived from the patent examples.
- Condense the difluoro-quinolone intermediate (II) with L-aminocaprolactam in acetonitrile using a base like triethylamine or pyridine to form the key intermediate (III).
- Purify intermediate (III) through standard workup procedures including washing with saturated NaCl and drying over anhydrous sodium sulfate.
- Perform late-stage chlorination on intermediate (III) using chlorosulfonic acid in chloroform under controlled temperature conditions to yield besifloxacin.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the shift to this novel synthetic route offers compelling advantages for procurement managers and supply chain directors focused on efficiency and margin protection. The primary driver of value is the substitution of the starting material. 2,4,5-Trifluorobenzoic acid is not only more chemically reactive, reducing processing time, but it is also generally more abundant and cost-effective in the global chemical market compared to its chlorinated counterpart. This raw material swap directly impacts the bill of materials (BOM), driving down the baseline cost of production without compromising quality. Additionally, the simplified purification requirements resulting from fewer side reactions mean reduced solvent consumption and lower waste disposal costs. These operational efficiencies compound to create a leaner manufacturing process that is more resilient to market fluctuations in raw material pricing.
- Cost Reduction in Manufacturing: The elimination of harsh reaction conditions and the reduction in reaction time significantly lower energy consumption and equipment wear. By avoiding the need for extensive chromatographic purification to remove chlorine-related impurities, manufacturers can save substantially on silica gel, solvents, and labor hours. The high yield reported in the final step further amplifies these savings by maximizing the output per batch, effectively spreading fixed costs over a larger volume of product. This logical deduction of cost benefits stems directly from the improved chemical efficiency of the fluoro-displacement mechanism.
- Enhanced Supply Chain Reliability: Utilizing a starting material that is easier to source and less prone to supply bottlenecks enhances the overall stability of the supply chain. The robustness of the reaction conditions means that the process is less sensitive to minor variations in temperature or reagent quality, leading to more consistent batch-to-batch performance. This reliability is crucial for meeting the stringent delivery schedules of major pharmaceutical clients. Furthermore, the stability of the intermediates allows for safer and more flexible logistics, reducing the risk of degradation during transport and storage.
- Scalability and Environmental Compliance: The streamlined nature of this synthesis makes it highly amenable to scale-up from pilot plant to commercial tonnage. Fewer unit operations and simpler workup procedures reduce the physical footprint required for production. From an environmental standpoint, the reduction in by-products and solvent usage aligns with green chemistry principles, helping manufacturers meet increasingly strict regulatory standards for waste discharge. The ability to produce high-purity high-purity pharmaceutical intermediates with a smaller environmental footprint is a significant competitive advantage in the modern regulatory landscape.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this besifloxacin synthesis route. These answers are derived directly from the experimental data and comparative analysis provided in the patent documentation. They serve to clarify the practical implications of adopting this technology for industrial production.
Q: Why is 2,4,5-trifluorobenzoic acid preferred over 5-chloro-2,4-difluorobenzoic acid?
A: The patent highlights that 2,4,5-trifluorobenzoic acid is significantly cheaper and more readily available. Furthermore, the fluorine atom at the reaction site is more reactive towards nucleophilic substitution than chlorine, allowing for milder reaction conditions and higher yields compared to traditional routes.
Q: How does this route ensure chiral purity?
A:
Q: What are the advantages of late-stage chlorination in this process?
A: Introducing the chlorine atom at the final step reduces the probability of side reactions that often occur when chlorine is present early in the synthesis. This strategic timing improves the overall reaction yield and simplifies the purification of the final API intermediate.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Besifloxacin Supplier
The technical advancements detailed in CN103044397A underscore the potential for optimizing besifloxacin production, yet translating patent chemistry into commercial reality requires specialized expertise. NINGBO INNO PHARMCHEM stands ready to bridge this gap, leveraging our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of intermediate meets the exacting standards required for ophthalmic formulations. We understand the critical nature of chiral purity and impurity control in fluoroquinolone synthesis, and our process engineers are adept at fine-tuning reaction parameters to maximize yield and quality.
We invite global partners to collaborate with us on optimizing their supply chains for besifloxacin and related intermediates. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage potential clients to reach out for specific COA data and route feasibility assessments to verify how this novel fluoro-substitution strategy can enhance your manufacturing economics. Let us partner to deliver high-quality, cost-effective solutions for the next generation of anti-infective therapies.