Advanced Industrial Synthesis of Entecavir: Overcoming Toxicity and Cost Barriers in Antiviral API Manufacturing
Advanced Industrial Synthesis of Entecavir: Overcoming Toxicity and Cost Barriers in Antiviral API Manufacturing
The global demand for effective antiviral therapies continues to drive innovation in the pharmaceutical intermediate sector, particularly for treatments targeting chronic Hepatitis B. Patent CN100379736C introduces a transformative preparation method for Entecavir, a potent deoxyguanosine analogue known for its high selectivity and low cytotoxicity. This technical insight report analyzes the proprietary synthesis route detailed in the patent, highlighting its strategic advantages over legacy methods. The core innovation lies in a streamlined four-step sequence that bypasses the use of hazardous reagents like boron trichloride, which have historically plagued the supply chain with safety and disposal challenges. By leveraging a modified Mitsunobu coupling followed by a precise oxidation-reduction sequence, this methodology offers a robust pathway for producing high-purity Entecavir suitable for rigorous regulatory standards.
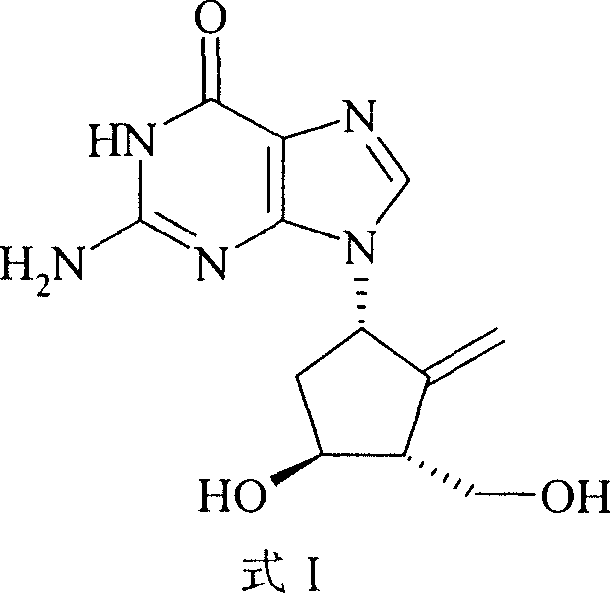
Entecavir functions by inhibiting the hepatitis B virus DNA polymerase and reverse transcriptase, exhibiting an antiviral activity significantly higher than its cytotoxicity profile. The structural integrity of the molecule, specifically the stereochemistry at the 1S, 3R, 4S positions and the exocyclic methylene group, is critical for its biological efficacy. Traditional synthesis routes often struggle to maintain this stereochemical purity while managing the economic burden of exotic reagents. The method disclosed in CN100379736C addresses these pain points directly by utilizing accessible starting materials and standard organic transformations. For R&D directors and procurement specialists, understanding this shift from complex, toxic chemistries to streamlined, safe processes is essential for securing a reliable supply of this critical antiviral agent in a competitive market landscape.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art technologies, such as those disclosed in Chinese patent ZL91110831.9 and international application WO98/09964, have long defined the baseline for Entecavir production but suffer from severe industrial drawbacks. These conventional pathways typically rely on the ring-opening of benzyl-protected epoxides with protected purines, followed by oxidation and carbene-like reactions to install the exocyclic double bond. A primary bottleneck in these legacy methods is the dependence on expensive chiral boron reagents as starting materials, which drastically inflates the raw material costs and limits supply chain flexibility. Furthermore, the final deprotection steps frequently necessitate the use of boron trichloride, a highly toxic and corrosive substance that poses significant safety risks to plant personnel and requires specialized, costly waste treatment infrastructure. The cumulative effect of these factors results in a lengthy synthesis with harsh reaction conditions, making the process economically unviable for large-scale commercialization and environmentally unsustainable.
The Novel Approach
In stark contrast, the novel approach presented in Patent CN100379736C重构 s the synthetic logic to prioritize safety, efficiency, and cost-effectiveness without compromising molecular integrity. This method initiates with a Mitsunobu reaction between a readily available cyclopentanol derivative and a protected purine base, establishing the crucial glycosidic bond under mild thermal conditions ranging from 0°C to 30°C. Instead of dangerous Lewis acids, the process employs a sequence of hydrolysis, periodate oxidation, and borohydride reduction to manipulate the side chain functionality, ensuring high selectivity and minimal byproduct formation. The installation of the exocyclic methylene group is achieved through a controlled tosylation and elimination strategy, avoiding the unpredictability of carbene chemistry. Finally, the deprotection is executed using methanolic hydrochloric acid followed by sodium in liquid ammonia, completely eliminating the need for boron trichloride. This paradigm shift not only simplifies the operational workflow but also aligns with modern green chemistry principles, offering a sustainable solution for API manufacturing.
Mechanistic Insights into Mitsunobu Coupling and Functional Group Manipulation
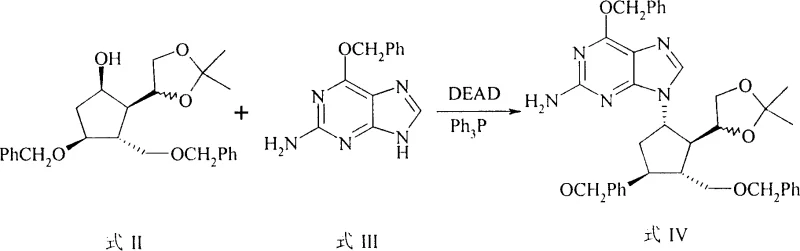
The cornerstone of this synthesis is the initial coupling step, which utilizes the Mitsunobu reaction to join the sugar mimic (Formula II) with the nucleobase (Formula III). Mechanistically, this involves the activation of the hydroxyl group on the cyclopentyl ring by triphenylphosphine and diethyl azodicarboxylate (DEAD), creating a reactive oxyphosphonium intermediate. This activation facilitates a nucleophilic attack by the N9 nitrogen of the purine base with inversion of configuration, ensuring the correct stereochemistry required for biological activity. The reaction is conducted in tetrahydrofuran at temperatures between 0°C and 30°C for 10 to 24 hours, conditions that are exceptionally mild compared to the high-energy inputs often required in traditional glycosylation. This precision control minimizes the formation of N7-isomers and other regioisomeric impurities, thereby simplifying downstream purification and enhancing the overall purity profile of the intermediate.
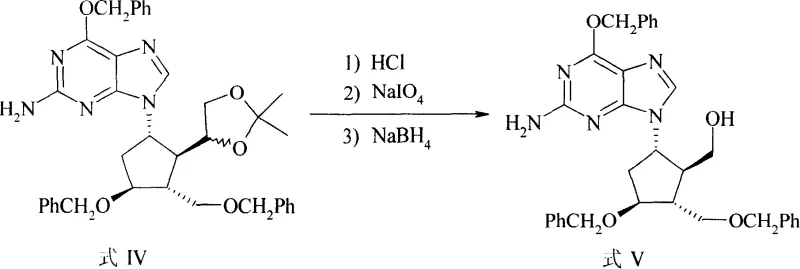
Following the coupling, the synthetic route proceeds through a sophisticated functional group interconversion sequence to prepare the ring for the final elimination. The acetal protection on the side chain is first hydrolyzed using concentrated hydrochloric acid, revealing a diol moiety that is subsequently subjected to oxidative cleavage by sodium periodate. This step selectively breaks the carbon-carbon bond to generate an aldehyde intermediate, which is immediately reduced in situ by sodium borohydride to a primary alcohol (Formula V). This one-pot oxidation-reduction strategy is highly efficient, preventing the accumulation of unstable aldehyde species that could lead to polymerization or side reactions. The subsequent dehydration involves converting the primary alcohol into a leaving group using p-toluenesulfonyl chloride, followed by base-induced elimination with sodium hydride. This E2 elimination mechanism cleanly generates the exocyclic double bond characteristic of Entecavir, demonstrating excellent regioselectivity and yielding the key precursor (Formula VI) with high fidelity.
How to Synthesize Entecavir Efficiently
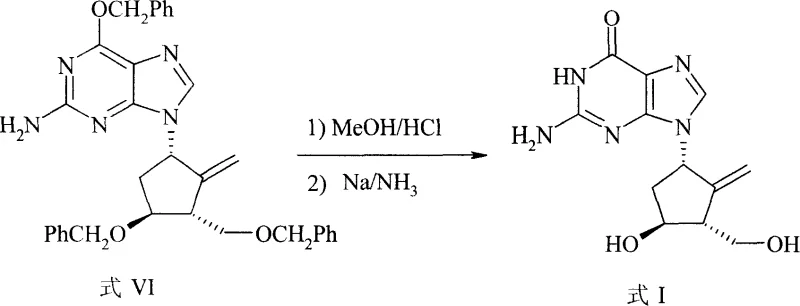
The synthesis of Entecavir via this patented route is designed for operational simplicity and high throughput, making it an ideal candidate for technology transfer and commercial scale-up. The process begins with the preparation of the coupled intermediate (Formula IV) through the aforementioned Mitsunobu protocol, followed by the sequential modification of the side chain to introduce the necessary hydroxymethyl and methylene functionalities. The final stage involves the global deprotection of the benzyl groups, a critical step that must be managed carefully to avoid hydrogenation of the sensitive exocyclic double bond. By utilizing dissolving metal reduction conditions (sodium in liquid ammonia) rather than catalytic hydrogenation, the method preserves the alkene integrity while efficiently cleaving the ether protections. Detailed standardized operating procedures for each transformation, including precise stoichiometric ratios and temperature controls, are essential to replicate the high yields reported in the patent examples.
- Perform Mitsunobu coupling between the protected cyclopentanol derivative and the purine base using triphenylphosphine and DEAD in THF at 0-30°C.
- Execute hydrolysis followed by oxidative cleavage with sodium periodate and reduction with sodium borohydride to modify the side chain.
- Conduct dehydration via tosylation and subsequent elimination with sodium hydride to form the exocyclic methylene group.
- Finalize the synthesis by removing protecting groups using methanolic HCl followed by dissolving metal reduction in liquid ammonia.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis route represents a significant opportunity to optimize cost structures and mitigate supply risks associated with Entecavir production. The elimination of exotic and hazardous reagents translates directly into reduced raw material expenditures and lower overhead costs related to safety compliance and waste management. By shifting away from dependencies on volatile chiral boron supplies, manufacturers can secure a more stable and predictable supply chain, insulating production schedules from market fluctuations of niche chemicals. Furthermore, the simplified operational parameters, such as ambient temperature reactions and standard solvent systems, reduce the energy consumption and equipment specialization required, facilitating easier integration into existing multi-purpose manufacturing facilities.
- Cost Reduction in Manufacturing: The most impactful economic advantage of this process is the complete removal of boron trichloride and expensive chiral boron starting materials from the bill of materials. Boron trichloride requires specialized handling equipment and rigorous neutralization protocols, the removal of which significantly lowers both capital expenditure and operational expenses. Additionally, the use of commodity chemicals like triphenylphosphine, sodium periodate, and tosyl chloride ensures that raw material sourcing is straightforward and cost-competitive. The high yields observed in the coupling step (approximately 83%) and the overall streamlined nature of the four-step sequence minimize material loss, further driving down the cost per kilogram of the final API.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the reliance on widely available, non-proprietary reagents that are produced by multiple global suppliers. Unlike legacy methods that might depend on single-source specialty reagents, this route utilizes standard organic building blocks that are less susceptible to geopolitical or logistical disruptions. The mild reaction conditions also reduce the risk of batch failures due to thermal runaway or equipment malfunction, ensuring consistent delivery timelines. This reliability is crucial for maintaining continuous production of antiviral medications, where interruptions can have significant public health implications.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, this method aligns perfectly with modern regulatory expectations for pharmaceutical manufacturing. The avoidance of heavy metals and highly toxic gases simplifies the effluent treatment process, reducing the environmental footprint of the facility. The reactions are performed in common solvents like tetrahydrofuran, methanol, and dichloromethane, which are easily recovered and recycled in a closed-loop system. This ease of waste management, combined with the robustness of the chemistry at scale, makes the process highly attractive for expansion from pilot plant to multi-ton commercial production without the need for extensive re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Entecavir synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in Patent CN100379736C, offering clarity on the practical benefits of adopting this novel route. Understanding these details is vital for stakeholders evaluating the feasibility of technology transfer or long-term supply agreements.
Q: How does this new method improve safety compared to traditional Entecavir synthesis?
A: Traditional methods often rely on highly toxic boron trichloride for deprotection and expensive chiral boron reagents. This patented process eliminates the need for boron trichloride, utilizing milder reagents like sodium in liquid ammonia and methanolic HCl, significantly reducing operational hazards and waste treatment costs.
Q: What are the typical yields for the key coupling step in this process?
A: According to the experimental data in Patent CN100379736C, the initial Mitsunobu coupling step (forming Formula IV) achieves a robust yield of approximately 83%, demonstrating high efficiency and suitability for large-scale production without significant material loss.
Q: Is this synthesis route suitable for commercial scale-up?
A: Yes, the process is explicitly designed for industrialization. It utilizes readily available raw materials, operates under mild temperature conditions (0-30°C for key steps), and avoids complex purification procedures, making it highly scalable for commercial API manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Entecavir Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and safe manufacturing processes in the pharmaceutical industry. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that complex syntheses like the Entecavir route described in CN100379736C are executed with precision. Our state-of-the-art facilities are equipped to handle the specific requirements of this chemistry, including low-temperature operations and dissolving metal reductions, while adhering to stringent purity specifications. With our rigorous QC labs and commitment to quality, we guarantee that every batch of Entecavir intermediate or API meets the highest international standards for safety and efficacy.
We invite potential partners to engage with our technical team to explore how this optimized synthesis route can benefit your specific supply chain needs. By collaborating with us, you gain access to a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating the tangible economic benefits of switching to this superior method. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, taking the first step towards a more reliable and cost-effective source of high-quality antiviral intermediates.