Optimizing Amlodipine Intermediate Production via Modified Hantzsch Cyclization for Commercial Scale
The pharmaceutical industry constantly seeks more efficient pathways for synthesizing critical cardiovascular agents, and the production of amlodipine intermediates remains a focal point for process optimization. Patent CN102070611A introduces a transformative approach to synthesizing the key dihydropyridine precursor, specifically 4-(2-chlorophenyl)-3-ethoxycarboxylic acid-5-methoxycarboxylic acid-6-methyl-2-(2-phthalamideethoxymethyl)-1,4-dihydropyridine. This innovation addresses long-standing inefficiencies in the classic Hantzsch pyridine synthesis by decoupling the reaction into a controlled two-step sequence rather than a chaotic one-pot mixture. By strategically employing a Knoevenagel condensation followed by a catalyzed cyclization, the methodology achieves a dramatic reduction in reaction time while simultaneously elevating product purity to approximately 99.9%. For R&D directors and process chemists, this represents a significant leap forward in managing impurity profiles and ensuring batch-to-batch consistency in complex API manufacturing.
Furthermore, the economic implications of this technical breakthrough extend deeply into supply chain management and cost structures. Traditional methods often struggled with yields hovering around 17.4% to 31.7%, necessitating massive over-processing of raw materials to achieve target output volumes. The new protocol described in the patent data demonstrates a robust total yield exceeding 68%, which fundamentally alters the cost basis for producing this high-value intermediate. By eliminating the need for hazardous azide reagents and reducing energy-intensive reflux times from roughly 20 hours down to a combined 7 to 8 hours, the process offers a compelling value proposition for procurement teams looking to secure reliable pharmaceutical intermediates supplier partnerships. This report will dissect the mechanistic advantages and commercial viability of adopting this refined synthetic route for global amlodipine production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of dihydropyridine rings for calcium channel blockers has relied heavily on the ternary Hantzsch reaction, where an aldehyde, a beta-keto ester, and an enamine are mixed simultaneously. As illustrated in the reaction scheme below, this conventional one-pot approach, while conceptually simple, suffers from severe thermodynamic and kinetic drawbacks that plague industrial scalability. The simultaneous presence of all three reactive components often leads to uncontrolled polymerization and the formation of intractable byproducts, resulting in notoriously low isolated yields. Specific prior art, such as the route depicted in Figure 1, reports yields as dismal as 17.4%, which is economically unsustainable for commercial API manufacturing where margin compression is a constant threat.
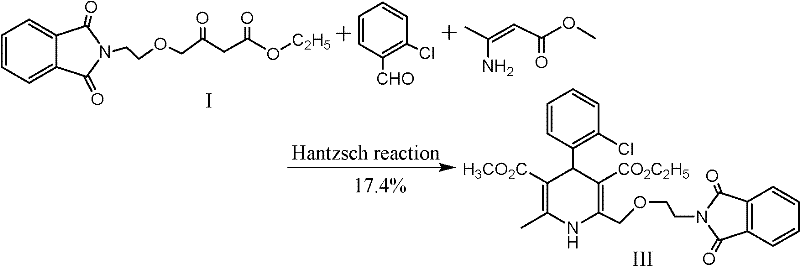
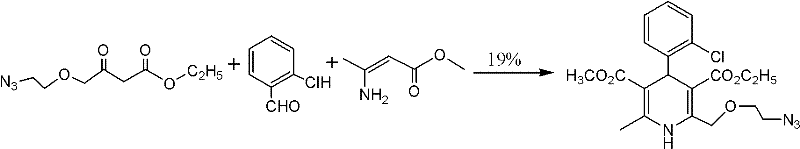
Beyond the issue of low yield, alternative historical pathways have introduced unacceptable safety risks and quality inconsistencies. For instance, certain Korean patents explored the use of azido groups as latent amino functionalities to construct the dihydropyridine core. However, as shown in Figure 2, this strategy not only resulted in poor yields of around 19% but also introduced the handling of unstable azide compounds, which pose explosion hazards and require specialized containment infrastructure that many standard chemical plants lack. Additionally, other methods utilizing ammonium acetate for cyclization, while achieving higher theoretical yields, have been plagued by poor reproducibility and inferior product appearance, making them unsuitable for the stringent quality standards required by regulatory bodies for cardiovascular medications.
The Novel Approach
The patented methodology fundamentally reengineers the synthesis by splitting the transformation into two distinct, optimized stages: a Knoevenagel condensation followed by a Hantzsch cyclization. This sequential approach allows for the precise control of reaction conditions at each stage, preventing the cross-reactivity that plagues the one-pot method. In the first stage, 4-[2-(phthalimido)ethoxy]ethyl acetoacetate reacts with o-chlorobenzaldehyde under the catalysis of acetic acid and piperidine. Crucially, the process employs cyclohexane as an azeotropic agent to continuously remove the water generated during condensation. This physical removal of the byproduct drives the equilibrium strongly towards the formation of the benzylidene intermediate (Intermediate II), ensuring near-quantitative conversion before the second step even begins.
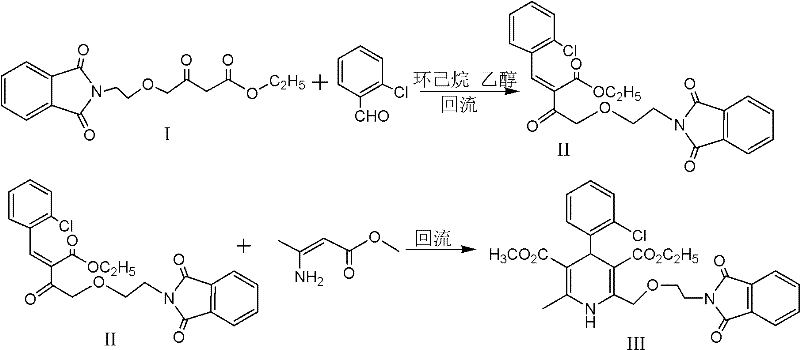
Once the benzylidene intermediate is formed, it undergoes cyclization with methyl 3-aminocrotonate in an alcoholic solvent catalyzed by an organic acid. This second step proceeds rapidly, typically completing within 5 to 6 hours, compared to the 20 hours required for traditional methods. The result is a highly pure dihydropyridine derivative with excellent crystal morphology and a total yield that can reach 68.6%. This telescoped yet controlled process eliminates the need for isolating the unstable Intermediate II, thereby reducing solvent usage and handling time. For a reliable pharmaceutical intermediates supplier, this translates to a streamlined workflow that minimizes waste generation and maximizes throughput, directly addressing the pain points of cost reduction in API manufacturing identified by procurement leaders.
Mechanistic Insights into Modified Hantzsch Cyclization
The success of this synthetic route lies in the mechanistic decoupling of the carbon-carbon bond formation from the ring-closing event. In the initial Knoevenagel condensation, the piperidine acts as a base to deprotonate the active methylene group of the acetoacetate derivative, generating a nucleophilic enolate. This enolate attacks the carbonyl carbon of the o-chlorobenzaldehyde, forming a beta-hydroxy intermediate which subsequently dehydrates to form the alpha,beta-unsaturated ketone (Intermediate II). The use of acetic acid alongside piperidine creates a buffered environment that facilitates proton transfer without promoting excessive side reactions. The continuous removal of water via the cyclohexane azeotrope is the thermodynamic driver here; by Le Chatelier's principle, removing the product (water) forces the reaction to completion, effectively suppressing the reverse hydrolysis reaction that often limits yield in closed systems.
In the subsequent cyclization phase, the mechanism shifts to a Michael addition followed by intramolecular condensation. The amino group of methyl 3-aminocrotonate attacks the beta-carbon of the benzylidene intermediate in a conjugate addition. The presence of the organic acid catalyst (acetic or propionic acid) activates the carbonyl groups and facilitates the necessary proton transfers for the final dehydration and aromatization steps that establish the 1,4-dihydropyridine ring. This acid-catalyzed environment is milder and more selective than the harsh conditions often required in uncatalyzed thermal cyclizations. By controlling the acidity and temperature (refluxing at 78-82°C), the process ensures that the phthalimide protecting group remains intact while the dihydropyridine core forms cleanly. This precise control over the reaction microenvironment is what allows the process to achieve the reported 99.9% purity, minimizing the formation of regioisomers or oxidized pyridine byproducts that are difficult to remove downstream.
How to Synthesize Amlodipine Intermediate Efficiently
The implementation of this synthesis requires careful attention to stoichiometry and water management to replicate the high yields reported in the patent literature. The process begins with the preparation of the benzylidene intermediate by refluxing the starting materials with a catalytic amount of piperidine acetate in a cyclohexane-ethanol mixture. Once the water evolution ceases, indicating complete condensation, the solvent may be partially removed or adjusted, and the enamine component is introduced directly into the same vessel. This "one-pot, two-step" telescoping strategy reduces unit operations and solvent consumption. The detailed standardized synthesis steps, including specific molar ratios and temperature profiles for scaling this reaction from laboratory to pilot plant, are outlined in the guide below.
- Perform Knoevenagel condensation between 4-[2-(phthalimido)ethoxy]ethyl acetoacetate and o-chlorobenzaldehyde using acetic acid and piperidine catalysts, removing water via cyclohexane reflux.
- Without isolating the intermediate, proceed directly to cyclization by adding methyl 3-aminocrotonate and an organic acid catalyst in an alcoholic solvent.
- Reflux the mixture for 5 to 6 hours, followed by vacuum distillation and crystallization using acetic acid to obtain the final intermediate with approximately 99.9% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented process offers tangible benefits that go beyond simple chemistry metrics. The primary advantage is the drastic reduction in processing time, which directly correlates to increased asset utilization and lower fixed costs per kilogram of product. By shortening the total reaction cycle from approximately 20 hours in legacy methods to roughly 7 hours, manufacturers can significantly increase their production capacity without investing in new reactor volume. This efficiency gain allows for more flexible scheduling and faster response times to market demand fluctuations, enhancing the overall reliability of the supply chain for critical cardiovascular medications. Furthermore, the elimination of hazardous azide reagents removes a major logistical and safety burden, simplifying regulatory compliance and reducing insurance and containment costs associated with explosive materials.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the substantial improvement in yield and the reduction in energy consumption. Increasing the yield from roughly 17% to nearly 69% means that less than one-third of the raw material input is required to produce the same amount of final product compared to older methods. This massive reduction in material waste directly lowers the variable cost of goods sold. Additionally, the shorter reaction times result in significantly lower utility costs for heating and stirring, while the ability to telescope the steps without isolating Intermediate II reduces solvent purchase and disposal expenses. These factors combine to create a much leaner cost structure, enabling competitive pricing strategies in the global generic drug market.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by processes that are sensitive to minor variations in conditions or rely on unstable intermediates. The robust nature of this two-step method, which utilizes stable, commodity-grade reagents like o-chlorobenzaldehyde and methyl 3-aminocrotonate, ensures consistent production outcomes. The high purity of the crude product (approx. 99.9%) reduces the burden on downstream purification steps, such as recrystallization or chromatography, which are often bottlenecks in API manufacturing. By minimizing the risk of batch failures due to side reactions or impurity buildup, suppliers can guarantee more reliable delivery schedules to their pharmaceutical clients, fostering stronger long-term partnerships.
- Scalability and Environmental Compliance: From an environmental and safety perspective, this route is inherently greener and safer than its predecessors. The avoidance of azides eliminates the risk of thermal runaway explosions, making the process safer to scale up to multi-ton quantities. The use of common solvents like ethanol and cyclohexane, which are easily recovered and recycled, aligns with modern green chemistry principles and reduces the environmental footprint of the manufacturing site. The high atom economy achieved through the high-yield condensation means less chemical waste is generated per unit of product, simplifying wastewater treatment and helping manufacturers meet increasingly stringent environmental regulations without costly end-of-pipe solutions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. Understanding these details is crucial for technical teams evaluating the feasibility of technology transfer. The answers provided are derived directly from the experimental data and comparative analysis found within the patent documentation, ensuring accuracy for decision-making purposes.
Q: How does the two-step Hantzsch method improve yield compared to traditional one-pot synthesis?
A: Traditional one-pot ternary reactions often suffer from low yields (around 17.4%) due to competitive side reactions and equilibrium limitations. The patented two-step method isolates the Knoevenagel condensation phase, driving it to completion by removing water, which significantly boosts the overall yield to over 68%.
Q: What are the safety advantages of avoiding azide-based routes for this intermediate?
A: Alternative synthetic routes utilizing azido groups as potential amino functionalities pose significant safety hazards due to the instability of azide compounds. The disclosed method avoids these hazardous reagents entirely, relying on stable amine precursors like methyl 3-aminocrotonate, making it far safer for large-scale industrial production.
Q: Why is water removal critical in the first step of this synthesis?
A: The initial Knoevenagel condensation is an equilibrium reaction that generates water as a byproduct. By using cyclohexane in a reflux setup with a water separator (Dean-Stark trap), the generated water is continuously removed, shifting the equilibrium towards the formation of the benzylidene intermediate and reducing reaction time from 8 hours to just 2 hours.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Amlodipine Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the efficiency of your final API production depends heavily on the quality and consistency of your starting materials. Our technical team has extensively analyzed advanced synthetic routes like the one described in CN102070611A to ensure our manufacturing capabilities align with the highest industry standards. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, allowing us to adapt quickly to process optimizations that enhance yield and purity. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of amlodipine intermediate meets the exacting requirements for cardiovascular drug formulation.
We invite global pharmaceutical partners to collaborate with us on optimizing their supply chains for dihydropyridine calcium antagonists. By leveraging our expertise in process chemistry and scale-up engineering, we can help you realize the full commercial potential of these improved synthetic methods. Please contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our high-purity amlodipine intermediates can drive value and stability in your manufacturing operations.