Optimizing Tulathromycin Synthesis: A Strategic Breakthrough in Intermediate Salt Purification for Commercial Scale
The pharmaceutical landscape for veterinary antibiotics is constantly evolving, driven by the need for higher purity standards and more efficient manufacturing processes that can withstand rigorous regulatory scrutiny. Patent CN111072730B introduces a significant advancement in the preparation of tulathromycin intermediate salts, specifically addressing the longstanding challenges associated with the purification of dihydrohomoerythromycin epoxide. This technology represents a pivotal shift from traditional isolation techniques, offering a robust pathway to achieve superior chemical integrity before the final amination step. By utilizing L-tartaric acid to form a stable intermediate salt, manufacturers can effectively bypass complex chromatographic separations that often bottleneck production capacity. This innovation is not merely a laboratory curiosity but a scalable solution designed for reliable agrochemical intermediate supplier networks seeking to optimize their value chains. The strategic implementation of this salt formation technique ensures that the molecular architecture of the macrolide ring remains intact while systematically removing structurally similar impurities that typically persist through standard workups. For industry stakeholders, this translates to a more predictable supply of high-purity OLED material precursors and veterinary actives, reinforcing the importance of adopting such refined synthetic methodologies in modern chemical manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of tulathromycin has relied on direct amination of crude dihydrohomoerythromycin epoxide, a process fraught with inefficiencies that compromise both yield and final product quality. Conventional routes often struggle with the presence of fermentation byproducts and incomplete reaction species that co-elute with the desired intermediate, leading to HPLC purities that hover around 80% or lower. These impurities act as nucleation inhibitors or participate in side reactions during the subsequent ring-opening amination, generating difficult-to-remove analogues that degrade the therapeutic profile of the final antibiotic. Furthermore, the lack of a distinct crystalline intermediate state forces reliance on extensive solvent exchanges and repetitive recrystallizations, which drastically increase operational expenditures and environmental waste loads. The thermal instability of the free base epoxide under prolonged heating conditions further exacerbates degradation, limiting the feasible batch sizes and complicating the commercial scale-up of complex polymer additives or pharmaceutical intermediates. Without a robust purification checkpoint at the epoxide stage, the cumulative effect of these impurities necessitates expensive downstream processing, eroding profit margins and extending lead times for high-purity pharmaceutical intermediates delivery to global markets.
The Novel Approach
The novel approach detailed in the patent data revolutionizes this workflow by introducing a targeted salt formation step using L-tartaric acid, which acts as a highly selective purification agent. By converting the free base epoxide into its L-tartrate salt, the process exploits specific solubility characteristics that favor the precipitation of the desired compound while leaving impurities in the mother liquor. This transformation is conducted in common organic solvents like acetone at moderate temperatures ranging from 40°C to 60°C, ensuring thermal stability while promoting rapid crystal growth. The resulting white solid suspension can be easily filtered, yielding an intermediate salt with purity levels exceeding 90%, a substantial improvement over the crude feedstock. This purified salt serves as a superior substrate for the final amination reaction, minimizing side reactions and streamlining the isolation of the final tulathromycin product. The simplicity of this post-treatment operation eliminates the need for complex extraction sequences, thereby enhancing the overall process mass intensity and aligning with green chemistry principles essential for modern cost reduction in electronic chemical manufacturing and pharmaceutical sectors alike.
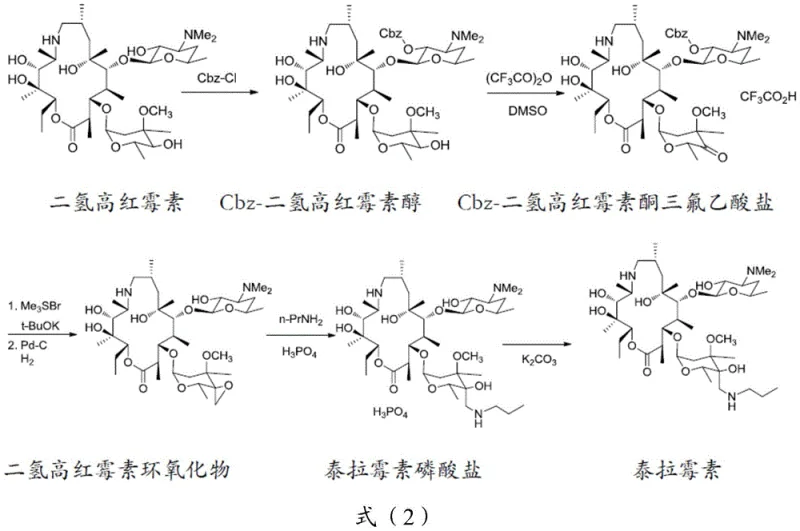
Mechanistically, the success of this purification strategy lies in the stereochemical complementarity between the chiral centers of the dihydrohomoerythromycin epoxide and the L-tartaric acid. The formation of the salt creates a rigid lattice structure that is thermodynamically more stable than the amorphous impurities present in the reaction mixture. As the solution cools from the reaction temperature of 50°C down to 0°C or even -10°C, the solubility product of the tartrate salt is exceeded, driving the selective crystallization of the target molecule. This process effectively rejects non-complementary isomers and degradation products, which remain solvated in the acetone or alcohol medium. The molar ratio of epoxide to tartaric acid is carefully controlled, typically around 1:1.1, to ensure complete conversion without excess acid contamination. This precise stoichiometric control is critical for maintaining the pH balance required for stable crystal growth, preventing the formation of oils that could trap impurities. The result is a highly ordered crystalline or semi-crystalline solid that exhibits consistent physical properties, facilitating reliable handling and storage prior to the next synthetic transformation.
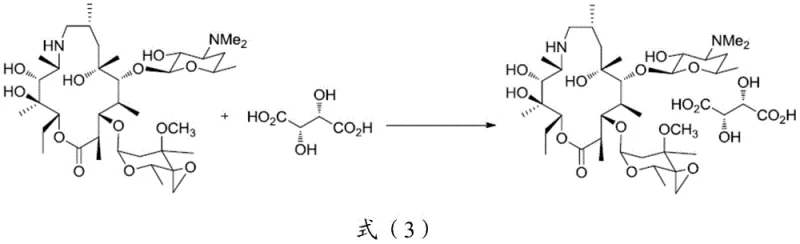
Impurity control is further enhanced by the kinetic selectivity of the crystallization process, which allows for the exclusion of closely related macrolide analogues that often plague erythromycin-derived syntheses. The hydrogen bonding network established within the tartrate salt lattice is specific to the spatial arrangement of hydroxyl and amino groups on the epoxide scaffold. Impurities lacking this precise configuration cannot integrate into the growing crystal face and are thus excluded from the solid phase. This phenomenon is akin to a molecular sieve effect achieved through thermodynamic equilibrium rather than physical filtration. Consequently, the HPLC profile of the isolated salt shows a marked reduction in peak area for known byproducts, simplifying the analytical burden on quality control laboratories. For R&D teams focused on purity and杂质谱 (impurity profiles), this mechanism offers a deterministic way to manage chemical risk, ensuring that the input material for the final amination step meets stringent specifications without requiring additional chromatographic polishing steps that are costly and time-consuming to validate.
How to Synthesize Dihydrohomoerythromycin Epoxide L-Tartrate Efficiently
To implement this advanced purification protocol effectively, manufacturers must adhere to precise operational parameters that govern the nucleation and growth of the intermediate salt crystals. The process begins with the dissolution of the crude epoxide in a selected organic solvent, with acetone being the preferred medium due to its favorable solubility gradient and ease of removal. Heating the mixture to 50°C ensures a homogeneous solution before the introduction of the resolving agent. The dropwise addition of L-tartaric acid must be managed carefully to avoid local supersaturation that could lead to amorphous precipitation rather than defined crystal growth. Once the addition is complete, a holding period at elevated temperature allows for Ostwald ripening, where smaller, less stable crystals dissolve and redeposit onto larger, more stable ones. The subsequent cooling phase is equally critical; a controlled ramp down to 0°C maximizes yield while maintaining purity. Detailed standardized synthesis steps see the guide below for exact procedural specifics tailored for industrial replication.
- Dissolve dihydrohomoerythromycin epoxide in an organic solvent such as acetone and heat the mixture to a temperature range of 40-60°C under continuous stirring to ensure complete solubilization.
- Slowly add a solution of L-tartaric acid in the same organic solvent dropwise to the reaction mixture, maintaining the temperature to initiate controlled nucleation and crystal growth of the intermediate salt.
- Cool the reaction suspension to a temperature between -10°C and 0°C to maximize precipitation, then filter the resulting white solid to isolate the high-purity dihydrohomoerythromycin epoxide L-tartrate.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this intermediate salt technology offers profound advantages that extend beyond mere chemical elegance. The primary benefit lies in the drastic simplification of the manufacturing workflow, which directly correlates to reduced operational costs and enhanced throughput. By eliminating the need for multiple extraction and washing steps traditionally required to purge impurities from the free base epoxide, facilities can significantly reduce solvent consumption and waste disposal fees. This streamlining of the process also shortens the overall cycle time per batch, allowing for greater production capacity within existing infrastructure constraints. For procurement managers, this means a more resilient supply base capable of responding quickly to market demand fluctuations without the risk of bottlenecks associated with complex purification trains. The robustness of the crystallization process also ensures consistent batch-to-batch quality, reducing the incidence of out-of-specification materials that can disrupt downstream formulation schedules and delay product launches.
- Cost Reduction in Manufacturing: The economic impact of this technology is driven by the elimination of expensive and time-consuming purification stages that characterize conventional synthesis routes. By achieving high purity through a single crystallization event, manufacturers avoid the capital and operational expenditures associated with preparative chromatography or repeated recrystallizations. The use of commodity solvents like acetone and ethanol further lowers raw material costs compared to specialized halogenated solvents often required for difficult separations. Additionally, the higher yield of the subsequent amination step, resulting from the cleaner intermediate, reduces the effective cost per kilogram of the final active pharmaceutical ingredient. These cumulative savings create a competitive pricing structure that allows suppliers to offer more attractive terms to long-term partners while maintaining healthy margins in a volatile market environment.
- Enhanced Supply Chain Reliability: Supply chain continuity is bolstered by the robustness and scalability of the salt formation process, which is less sensitive to minor variations in feedstock quality compared to direct amination methods. The ability to store the stable intermediate salt provides a strategic buffer, allowing manufacturers to decouple the upstream fermentation or synthesis of the epoxide from the final drug substance production. This inventory flexibility mitigates risks associated with equipment downtime or raw material shortages, ensuring a steady flow of materials to customers. Furthermore, the simplified process requires less specialized operator training and reduces the likelihood of human error during complex workup procedures. For supply chain heads, this translates to reduced lead time for high-purity pharmaceutical intermediates and a more predictable delivery schedule, which is crucial for maintaining just-in-time manufacturing models in the global veterinary pharmaceutical sector.
- Scalability and Environmental Compliance: The environmental footprint of the manufacturing process is significantly reduced through the minimization of solvent usage and waste generation inherent in this streamlined approach. Crystallization is inherently a greener unit operation compared to extraction-heavy workflows, as it relies on phase changes rather than bulk solvent partitioning. The recovery of solvents like acetone and ethanol is straightforward and energy-efficient, aligning with increasingly stringent environmental regulations and corporate sustainability goals. The process is also readily scalable from pilot plant to commercial production volumes without the need for specialized equipment, facilitating rapid technology transfer between sites. This scalability ensures that the benefits of the innovation can be realized across the entire production network, supporting the commercial scale-up of complex veterinary drugs while adhering to global standards for environmental stewardship and safety compliance.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this tulathromycin intermediate synthesis technology. These insights are derived directly from the patent specifications and practical considerations for industrial application, providing clarity on how this method compares to legacy processes. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios. The answers reflect a commitment to transparency and technical accuracy, ensuring that decision-makers have the necessary information to assess the potential impact on their operations and product quality standards.
Q: How does the L-tartaric acid salt formation improve intermediate purity compared to conventional methods?
A: The formation of the L-tartrate salt leverages specific solubility differences to exclude impurities during crystallization, raising HPLC purity from approximately 80% in crude epoxide to over 90% in the isolated salt, significantly simplifying downstream purification.
Q: What are the critical reaction conditions for scaling this intermediate synthesis?
A: Critical parameters include maintaining the reaction temperature between 40-60°C during acid addition to prevent oiling out, followed by precise cooling to 0°C or below to ensure high recovery yields of the crystalline salt suitable for industrial filtration.
Q: Does this purification method impact the final yield of Tulathromycin?
A: Yes, by starting with a higher purity intermediate (>90%), the subsequent amination step proceeds with fewer side reactions, resulting in a crude tulathromycin purity exceeding 96% and reducing the burden on final recrystallization steps.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tulathromycin Intermediate Salt Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic routes like the L-tartrate salt formation to maintain competitiveness in the global veterinary pharmaceutical market. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless and efficient. We are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of intermediate meets the high standards required for downstream API synthesis. Our commitment to technical excellence allows us to navigate the complexities of macrolide chemistry with precision, delivering materials that facilitate smoother regulatory filings and faster time-to-market for our clients' finished products.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis route can be tailored to your specific supply chain requirements. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the potential economic benefits and operational efficiencies available through our partnership. We encourage you to reach out for specific COA data and route feasibility assessments that demonstrate our capability to support your long-term strategic goals. Let us collaborate to engineer a supply chain that is not only cost-effective but also resilient and capable of meeting the evolving demands of the international healthcare industry.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →